 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBenchmarking Pretrained Molecular Embedding Models For Molecular Representation Learning
Aug 08, 2025Pretrained neural networks have attracted significant interest in chemistry and small molecule drug design. Embeddings from these models are widely used for molecular property prediction, virtual screening, and small data learning in molecular chemistry. This study presents the most extensive comparison of such models to date, evaluating 25 models across 25 datasets. Under a fair comparison framework, we assess models spanning various modalities, architectures, and pretraining strategies. Using a dedicated hierarchical Bayesian statistical testing model, we arrive at a surprising result: nearly all neural models show negligible or no improvement over the baseline ECFP molecular fingerprint. Only the CLAMP model, which is also based on molecular fingerprints, performs statistically significantly better than the alternatives. These findings raise concerns about the evaluation rigor in existing studies. We discuss potential causes, propose solutions, and offer practical recommendations.
Evaluating machine learning models for predicting pesticides toxicity to honey bees
Apr 01, 2025Small molecules play a critical role in the biomedical, environmental, and agrochemical domains, each with distinct physicochemical requirements and success criteria. Although biomedical research benefits from extensive datasets and established benchmarks, agrochemical data remain scarce, particularly with respect to species-specific toxicity. This work focuses on ApisTox, the most comprehensive dataset of experimentally validated chemical toxicity to the honey bee (Apis mellifera), an ecologically vital pollinator. We evaluate ApisTox using a diverse suite of machine learning approaches, including molecular fingerprints, graph kernels, and graph neural networks, as well as pretrained models. Comparative analysis with medicinal datasets from the MoleculeNet benchmark reveals that ApisTox represents a distinct chemical space. Performance degradation on non-medicinal datasets, such as ApisTox, demonstrates their limited generalizability of current state-of-the-art algorithms trained solely on biomedical data. Our study highlights the need for more diverse datasets and for targeted model development geared toward the agrochemical domain.
Molecular Fingerprints Are Strong Models for Peptide Function Prediction
Jan 29, 2025
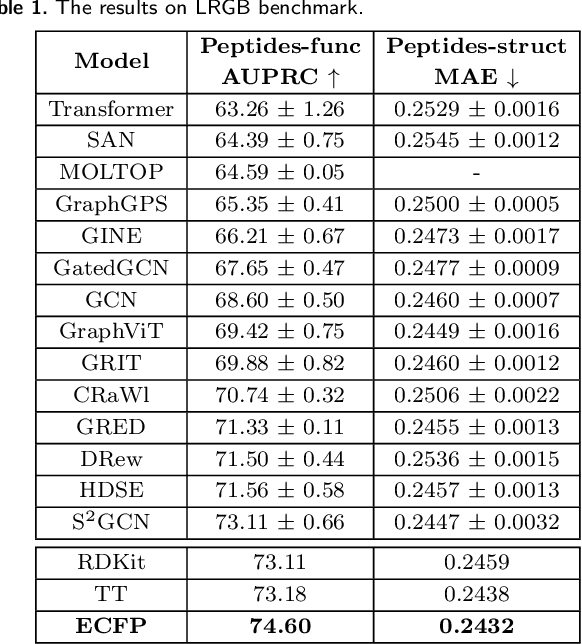

We study the effectiveness of molecular fingerprints for peptide property prediction and demonstrate that domain-specific feature extraction from molecular graphs can outperform complex and computationally expensive models such as GNNs, pretrained sequence-based transformers and multimodal ensembles, even without hyperparameter tuning. To this end, we perform a thorough evaluation on 126 datasets, achieving state-of-the-art results on LRGB and 5 other peptide function prediction benchmarks. We show that models based on count variants of ECFP, Topological Torsion, and RDKit molecular fingerprints and LightGBM as classification head are remarkably robust. The strong performance of molecular fingerprints, which are intrinsically very short-range feature encoders, challenges the presumed importance of long-range interactions in peptides. Our conclusion is that the use of molecular fingerprints for larger molecules, such as peptides, can be a computationally feasible, low-parameter, and versatile alternative to sophisticated deep learning models.
Scikit-fingerprints: easy and efficient computation of molecular fingerprints in Python
Jul 18, 2024



In this work, we present \textit{scikit-fingerprints}, a Python package for computation of molecular fingerprints for applications in chemoinformatics. Our library offers an industry-standard scikit-learn interface, allowing intuitive usage and easy integration with machine learning pipelines. It is also highly optimized, featuring parallel computation that enables efficient processing of large molecular datasets. Currently, \textit{scikit-fingerprints} stands as the most feature-rich library in the Python ecosystem, offering over 30 molecular fingerprints. Our library simplifies chemoinformatics tasks based on molecular fingerprints, including molecular property prediction and virtual screening. It is also flexible, highly efficient, and fully open source.
Molecular Topological Profile (MOLTOP) -- Simple and Strong Baseline for Molecular Graph Classification
Jul 16, 2024We revisit the effectiveness of topological descriptors for molecular graph classification and design a simple, yet strong baseline. We demonstrate that a simple approach to feature engineering - employing histogram aggregation of edge descriptors and one-hot encoding for atomic numbers and bond types - when combined with a Random Forest classifier, can establish a strong baseline for Graph Neural Networks (GNNs). The novel algorithm, Molecular Topological Profile (MOLTOP), integrates Edge Betweenness Centrality, Adjusted Rand Index and SCAN Structural Similarity score. This approach proves to be remarkably competitive when compared to modern GNNs, while also being simple, fast, low-variance and hyperparameter-free. Our approach is rigorously tested on MoleculeNet datasets using fair evaluation protocol provided by Open Graph Benchmark. We additionally show out-of-domain generation capabilities on peptide classification task from Long Range Graph Benchmark. The evaluations across eleven benchmark datasets reveal MOLTOP's strong discriminative capabilities, surpassing the $1$-WL test and even $3$-WL test for some classes of graphs. Our conclusion is that descriptor-based baselines, such as the one we propose, are still crucial for accurately assessing advancements in the GNN domain.
ApisTox: a new benchmark dataset for the classification of small molecules toxicity on honey bees
Apr 24, 2024The global decline in bee populations poses significant risks to agriculture, biodiversity, and environmental stability. To bridge the gap in existing data, we introduce ApisTox, a comprehensive dataset focusing on the toxicity of pesticides to honey bees (Apis mellifera). This dataset combines and leverages data from existing sources such as ECOTOX and PPDB, providing an extensive, consistent, and curated collection that surpasses the previous datasets. ApisTox incorporates a wide array of data, including toxicity levels for chemicals, details such as time of their publication in literature, and identifiers linking them to external chemical databases. This dataset may serve as an important tool for environmental and agricultural research, but also can support the development of policies and practices aimed at minimizing harm to bee populations. Finally, ApisTox offers a unique resource for benchmarking molecular property prediction methods on agrochemical compounds, facilitating advancements in both environmental science and cheminformatics. This makes it a valuable tool for both academic research and practical applications in bee conservation.
Strengthening structural baselines for graph classification using Local Topological Profile
May 01, 2023We present the analysis of the topological graph descriptor Local Degree Profile (LDP), which forms a widely used structural baseline for graph classification. Our study focuses on model evaluation in the context of the recently developed fair evaluation framework, which defines rigorous routines for model selection and evaluation for graph classification, ensuring reproducibility and comparability of the results. Based on the obtained insights, we propose a new baseline algorithm called Local Topological Profile (LTP), which extends LDP by using additional centrality measures and local vertex descriptors. The new approach provides the results outperforming or very close to the latest GNNs for all datasets used. Specifically, state-of-the-art results were obtained for 4 out of 9 benchmark datasets. We also consider computational aspects of LDP-based feature extraction and model construction to propose practical improvements affecting execution speed and scalability. This allows for handling modern, large datasets and extends the portfolio of benchmarks used in graph representation learning. As the outcome of our work, we obtained LTP as a simple to understand, fast and scalable, still robust baseline, capable of outcompeting modern graph classification models such as Graph Isomorphism Network (GIN). We provide open-source implementation at \href{https://github.com/j-adamczyk/LTP}{GitHub}.
Application of Graph Neural Networks and graph descriptors for graph classification
Nov 07, 2022Graph classification is an important area in both modern research and industry. Multiple applications, especially in chemistry and novel drug discovery, encourage rapid development of machine learning models in this area. To keep up with the pace of new research, proper experimental design, fair evaluation, and independent benchmarks are essential. Design of strong baselines is an indispensable element of such works. In this thesis, we explore multiple approaches to graph classification. We focus on Graph Neural Networks (GNNs), which emerged as a de facto standard deep learning technique for graph representation learning. Classical approaches, such as graph descriptors and molecular fingerprints, are also addressed. We design fair evaluation experimental protocol and choose proper datasets collection. This allows us to perform numerous experiments and rigorously analyze modern approaches. We arrive to many conclusions, which shed new light on performance and quality of novel algorithms. We investigate application of Jumping Knowledge GNN architecture to graph classification, which proves to be an efficient tool for improving base graph neural network architectures. Multiple improvements to baseline models are also proposed and experimentally verified, which constitutes an important contribution to the field of fair model comparison.