 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular Fingerprints Are Strong Models for Peptide Function Prediction
Jan 29, 2025
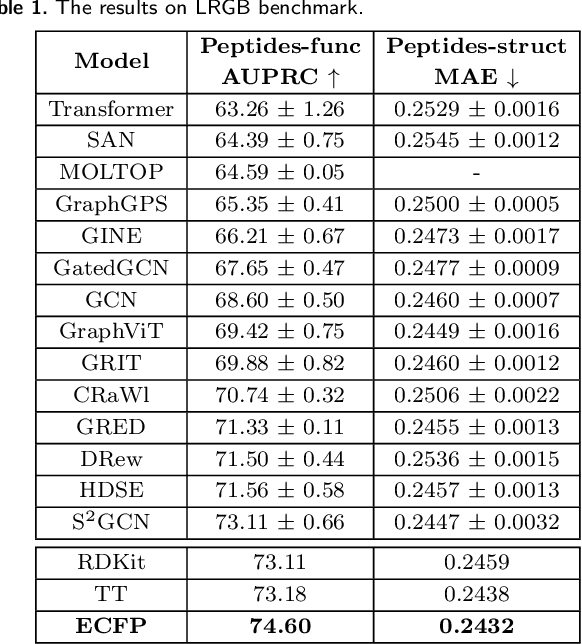
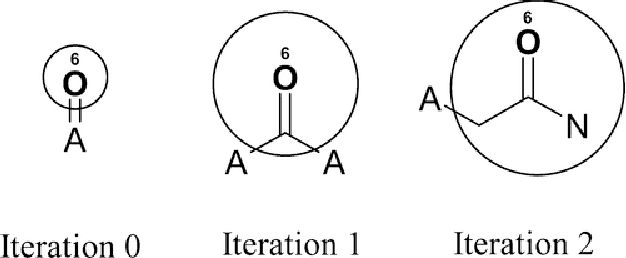

We study the effectiveness of molecular fingerprints for peptide property prediction and demonstrate that domain-specific feature extraction from molecular graphs can outperform complex and computationally expensive models such as GNNs, pretrained sequence-based transformers and multimodal ensembles, even without hyperparameter tuning. To this end, we perform a thorough evaluation on 126 datasets, achieving state-of-the-art results on LRGB and 5 other peptide function prediction benchmarks. We show that models based on count variants of ECFP, Topological Torsion, and RDKit molecular fingerprints and LightGBM as classification head are remarkably robust. The strong performance of molecular fingerprints, which are intrinsically very short-range feature encoders, challenges the presumed importance of long-range interactions in peptides. Our conclusion is that the use of molecular fingerprints for larger molecules, such as peptides, can be a computationally feasible, low-parameter, and versatile alternative to sophisticated deep learning models.
Scikit-fingerprints: easy and efficient computation of molecular fingerprints in Python
Jul 18, 2024



In this work, we present \textit{scikit-fingerprints}, a Python package for computation of molecular fingerprints for applications in chemoinformatics. Our library offers an industry-standard scikit-learn interface, allowing intuitive usage and easy integration with machine learning pipelines. It is also highly optimized, featuring parallel computation that enables efficient processing of large molecular datasets. Currently, \textit{scikit-fingerprints} stands as the most feature-rich library in the Python ecosystem, offering over 30 molecular fingerprints. Our library simplifies chemoinformatics tasks based on molecular fingerprints, including molecular property prediction and virtual screening. It is also flexible, highly efficient, and fully open source.
A Python library for efficient computation of molecular fingerprints
Mar 27, 2024
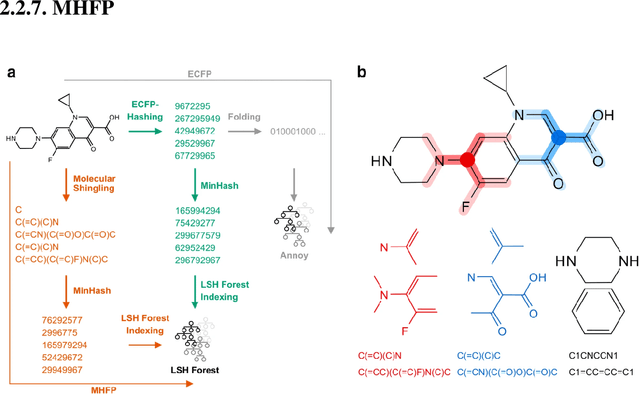
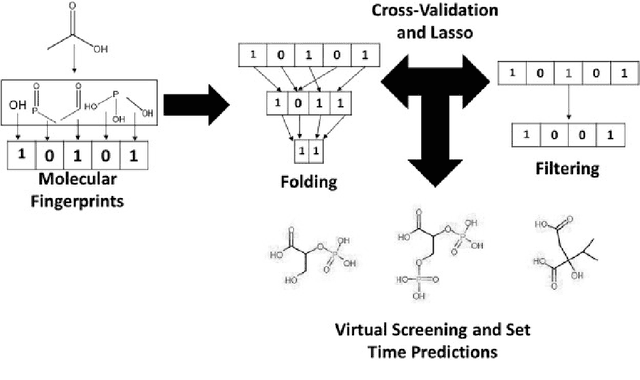
Machine learning solutions are very popular in the field of chemoinformatics, where they have numerous applications, such as novel drug discovery or molecular property prediction. Molecular fingerprints are algorithms commonly used for vectorizing chemical molecules as a part of preprocessing in this kind of solution. However, despite their popularity, there are no libraries that implement them efficiently for large datasets, utilizing modern, multicore architectures. On top of that, most of them do not provide the user with an intuitive interface, or one that would be compatible with other machine learning tools. In this project, we created a Python library that computes molecular fingerprints efficiently and delivers an interface that is comprehensive and enables the user to easily incorporate the library into their existing machine learning workflow. The library enables the user to perform computation on large datasets using parallelism. Because of that, it is possible to perform such tasks as hyperparameter tuning in a reasonable time. We describe tools used in implementation of the library and asses its time performance on example benchmark datasets. Additionally, we show that using molecular fingerprints we can achieve results comparable to state-of-the-art ML solutions even with very simple models.