 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEvaluating machine learning models for predicting pesticides toxicity to honey bees
Apr 01, 2025Small molecules play a critical role in the biomedical, environmental, and agrochemical domains, each with distinct physicochemical requirements and success criteria. Although biomedical research benefits from extensive datasets and established benchmarks, agrochemical data remain scarce, particularly with respect to species-specific toxicity. This work focuses on ApisTox, the most comprehensive dataset of experimentally validated chemical toxicity to the honey bee (Apis mellifera), an ecologically vital pollinator. We evaluate ApisTox using a diverse suite of machine learning approaches, including molecular fingerprints, graph kernels, and graph neural networks, as well as pretrained models. Comparative analysis with medicinal datasets from the MoleculeNet benchmark reveals that ApisTox represents a distinct chemical space. Performance degradation on non-medicinal datasets, such as ApisTox, demonstrates their limited generalizability of current state-of-the-art algorithms trained solely on biomedical data. Our study highlights the need for more diverse datasets and for targeted model development geared toward the agrochemical domain.
Detection of protein-ligand binding sites with 3D segmentation
Apr 13, 2019



In recent years machine learning (ML) took bio- and cheminformatics fields by storm, providing new solutions for a vast repertoire of problems related to protein sequence, structure, and interactions analysis. ML techniques, deep neural networks especially, were proven more effective than classical models for tasks like predicting binding affinity for molecular complex. In this work we investigated the earlier stage of drug discovery process - finding druggable pockets on protein surface, that can be later used to design active molecules. For this purpose we developed a 3D fully convolutional neural network capable of binding site segmentation. Our solution has high prediction accuracy and provides intuitive representations of the results, which makes it easy to incorporate into drug discovery projects. The model's source code, together with scripts for most common use-cases is freely available at http://gitlab.com/cheminfIBB/kalasanty
Development and evaluation of a deep learning model for protein-ligand binding affinity prediction
Jan 03, 2018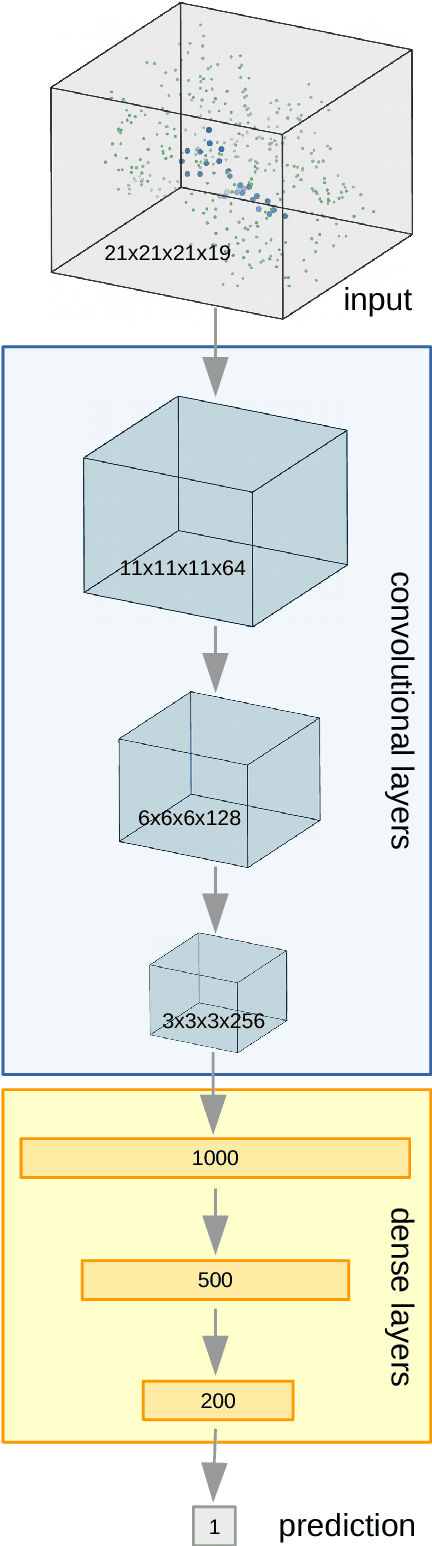
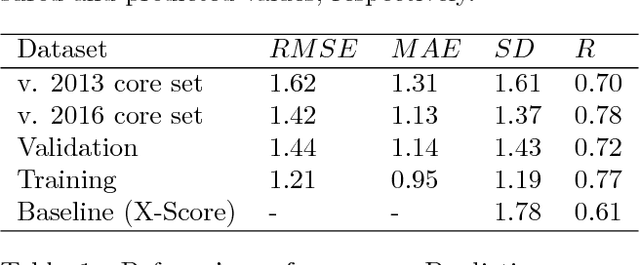


Structure based ligand discovery is one of the most successful approaches for augmenting the drug discovery process. Currently, there is a notable shift towards machine learning (ML) methodologies to aid such procedures. Deep learning has recently gained considerable attention as it allows the model to "learn" to extract features that are relevant for the task at hand. We have developed a novel deep neural network estimating the binding affinity of ligand-receptor complexes. The complex is represented with a 3D grid, and the model utilizes a 3D convolution to produce a feature map of this representation, treating the atoms of both proteins and ligands in the same manner. Our network was tested on the CASF "scoring power" benchmark and Astex Diverse Set and outperformed classical scoring functions. The model, together with usage instructions and examples, is available as a git repository at http://gitlab.com/cheminfIBB/pafnucy