 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDifferentiable Scaffolding Tree for Molecular Optimization
Sep 22, 2021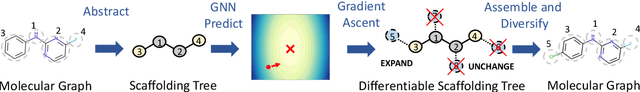

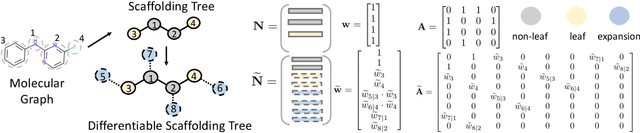
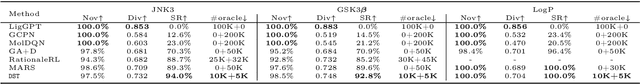
The structural design of functional molecules, also called molecular optimization, is an essential chemical science and engineering task with important applications, such as drug discovery. Deep generative models and combinatorial optimization methods achieve initial success but still struggle with directly modeling discrete chemical structures and often heavily rely on brute-force enumeration. The challenge comes from the discrete and non-differentiable nature of molecule structures. To address this, we propose differentiable scaffolding tree (DST) that utilizes a learned knowledge network to convert discrete chemical structures to locally differentiable ones. DST enables a gradient-based optimization on a chemical graph structure by back-propagating the derivatives from the target properties through a graph neural network (GNN). Our empirical studies show the gradient-based molecular optimizations are both effective and sample efficient. Furthermore, the learned graph parameters can also provide an explanation that helps domain experts understand the model output.