 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGSAE: an autoencoder with embedded gene-set nodes for genomics functional characterization
May 21, 2018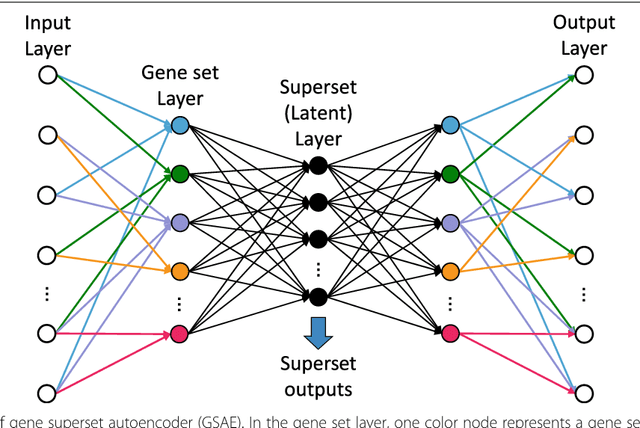
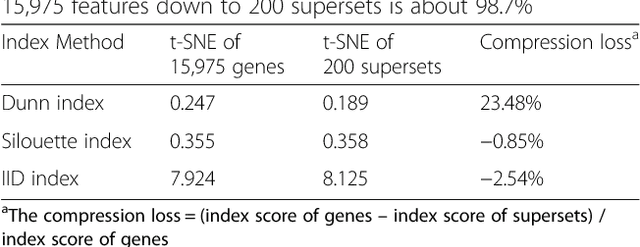
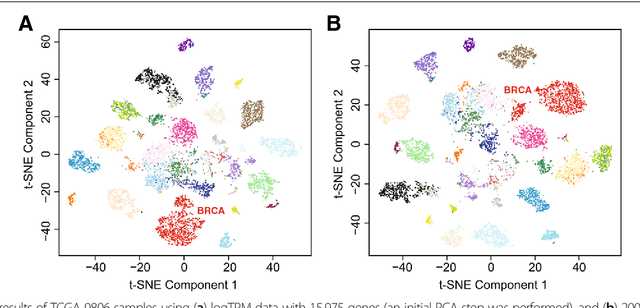
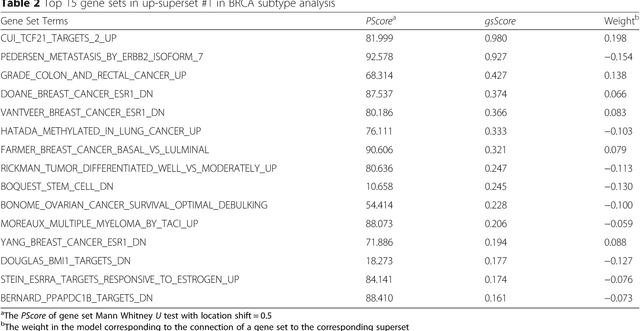
Bioinformatics tools have been developed to interpret gene expression data at the gene set level, and these gene set based analyses improve the biologists' capability to discover functional relevance of their experiment design. While elucidating gene set individually, inter gene sets association is rarely taken into consideration. Deep learning, an emerging machine learning technique in computational biology, can be used to generate an unbiased combination of gene set, and to determine the biological relevance and analysis consistency of these combining gene sets by leveraging large genomic data sets. In this study, we proposed a gene superset autoencoder (GSAE), a multi-layer autoencoder model with the incorporation of a priori defined gene sets that retain the crucial biological features in the latent layer. We introduced the concept of the gene superset, an unbiased combination of gene sets with weights trained by the autoencoder, where each node in the latent layer is a superset. Trained with genomic data from TCGA and evaluated with their accompanying clinical parameters, we showed gene supersets' ability of discriminating tumor subtypes and their prognostic capability. We further demonstrated the biological relevance of the top component gene sets in the significant supersets. Using autoencoder model and gene superset at its latent layer, we demonstrated that gene supersets retain sufficient biological information with respect to tumor subtypes and clinical prognostic significance. Superset also provides high reproducibility on survival analysis and accurate prediction for cancer subtypes.
Predicting drug response of tumors from integrated genomic profiles by deep neural networks
May 20, 2018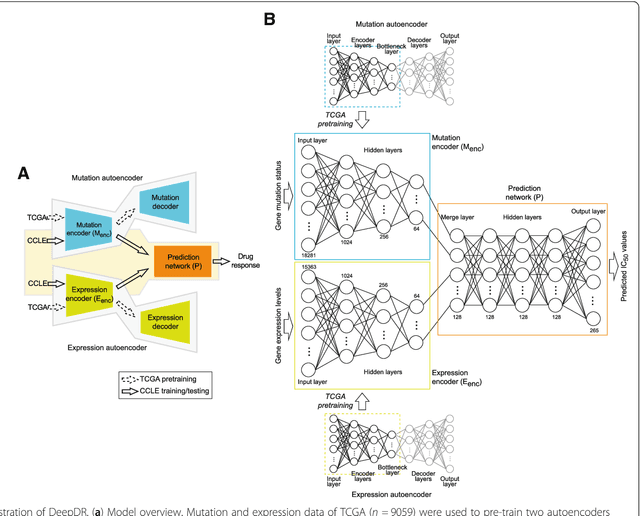
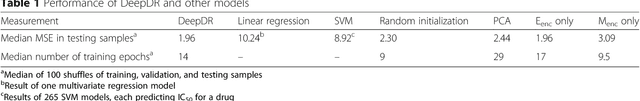

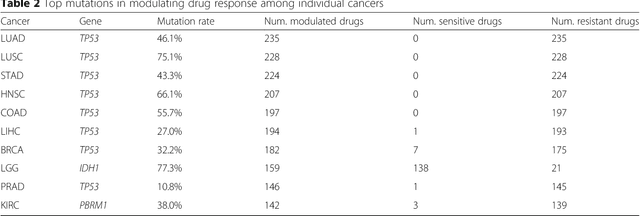
The study of high-throughput genomic profiles from a pharmacogenomics viewpoint has provided unprecedented insights into the oncogenic features modulating drug response. A recent screening of ~1,000 cancer cell lines to a collection of anti-cancer drugs illuminated the link between genotypes and vulnerability. However, due to essential differences between cell lines and tumors, the translation into predicting drug response in tumors remains challenging. Here we proposed a DNN model to predict drug response based on mutation and expression profiles of a cancer cell or a tumor. The model contains a mutation and an expression encoders pre-trained using a large pan-cancer dataset to abstract core representations of high-dimension data, followed by a drug response predictor network. Given a pair of mutation and expression profiles, the model predicts IC50 values of 265 drugs. We trained and tested the model on a dataset of 622 cancer cell lines and achieved an overall prediction performance of mean squared error at 1.96 (log-scale IC50 values). The performance was superior in prediction error or stability than two classical methods and four analog DNNs of our model. We then applied the model to predict drug response of 9,059 tumors of 33 cancer types. The model predicted both known, including EGFR inhibitors in non-small cell lung cancer and tamoxifen in ER+ breast cancer, and novel drug targets. The comprehensive analysis further revealed the molecular mechanisms underlying the resistance to a chemotherapeutic drug docetaxel in a pan-cancer setting and the anti-cancer potential of a novel agent, CX-5461, in treating gliomas and hematopoietic malignancies. Overall, our model and findings improve the prediction of drug response and the identification of novel therapeutic options.