 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAssessing the Reproducibility of Machine-learning-based Biomarker Discovery in Parkinson's Disease
Apr 06, 2023Genome-Wide Association Studies (GWAS) help identify genetic variations in people with diseases such as Parkinson's disease (PD), which are less common in those without the disease. Thus, GWAS data can be used to identify genetic variations associated with the disease. Feature selection and machine learning approaches can be used to analyze GWAS data and identify potential disease biomarkers. However, GWAS studies have technical variations that affect the reproducibility of identified biomarkers, such as differences in genotyping platforms and selection criteria for individuals to be genotyped. To address this issue, we collected five GWAS datasets from the database of Genotypes and Phenotypes (dbGaP) and explored several data integration strategies. We evaluated the agreement among different strategies in terms of the Single Nucleotide Polymorphisms (SNPs) that were identified as potential PD biomarkers. Our results showed a low concordance of biomarkers discovered using different datasets or integration strategies. However, we identified fifty SNPs that were identified at least twice, which could potentially serve as novel PD biomarkers. These SNPs are indirectly linked to PD in the literature but have not been directly associated with PD before. These findings open up new potential avenues of investigation.
Clustering, multicollinearity, and singular vectors
Aug 07, 2020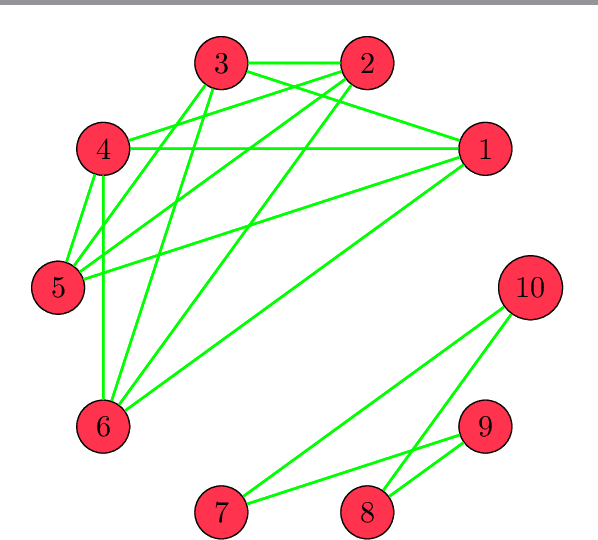
Let $A$ be a matrix with its pseudo-matrix $A^{\dagger}$ and set $S=I-A^{\dagger}A$. We prove that, after re-ordering the columns of $A$, the matrix $S$ has a block-diagonal form where each block corresponds to a set of linearly dependent columns. This allows us to identify redundant columns in $A$. We explore some applications in supervised and unsupervised learning, specially feature selection, clustering, and sensitivity of solutions of least squares solutions.
Detecting ulcerative colitis from colon samples using efficient feature selection and machine learning
Aug 04, 2020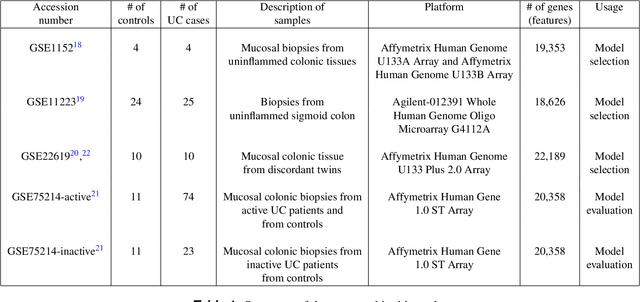
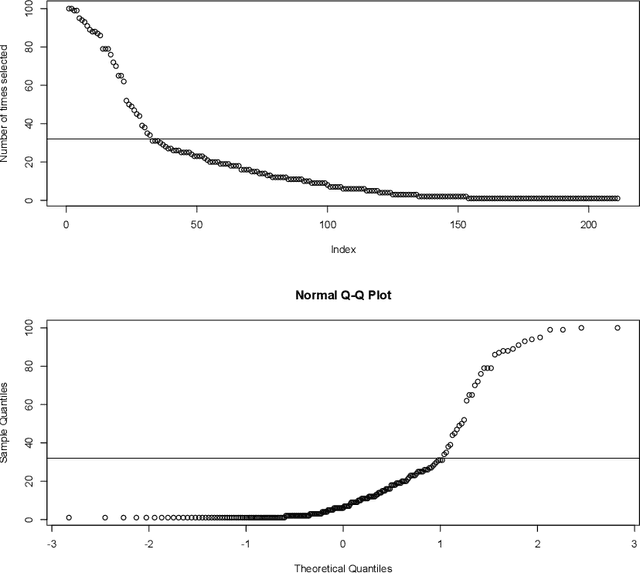

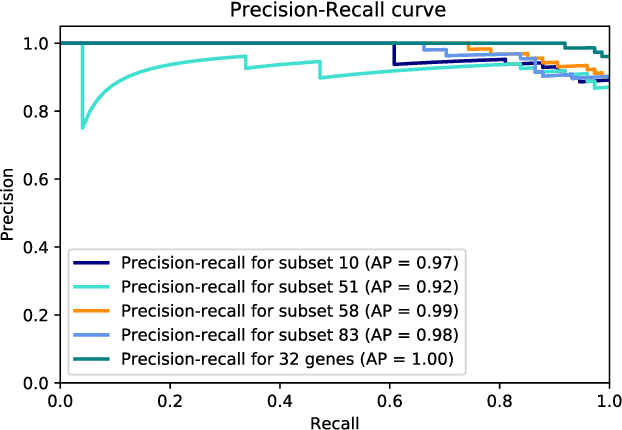
Ulcerative colitis (UC) is one of the most common forms of inflammatory bowel disease (IBD) characterized by inflammation of the mucosal layer of the colon. Diagnosis of UC is based on clinical symptoms, and then confirmed based on endoscopic, histologic and laboratory findings. Feature selection and machine learning have been previously used for creating models to facilitate the diagnosis of certain diseases. In this work, we used a recently developed feature selection algorithm (DRPT) combined with a support vector machine (SVM) classifier to generate a model to discriminate between healthy subjects and subjects with UC based on the expression values of 32 genes in colon samples. We validated our model with an independent gene expression dataset of colonic samples from subjects in active and inactive periods of UC. Our model perfectly detected all active cases and had an average precision of 0.62 in the inactive cases. Compared with results reported in previous studies and a model generated by a recently published software for biomarker discovery using machine learning (BioDiscML), our final model for detecting UC shows better performance in terms of average precision.
High-Dimensional Feature Selection for Genomic Datasets
Feb 27, 2020
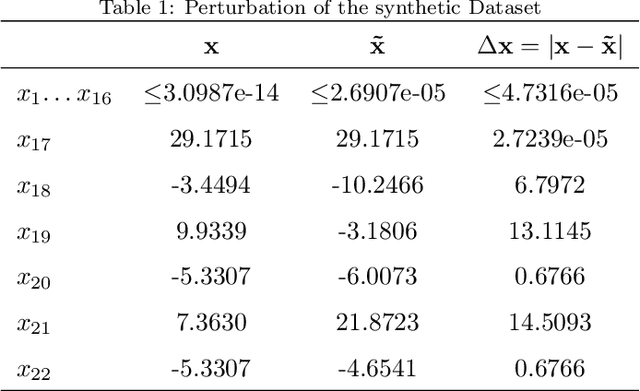
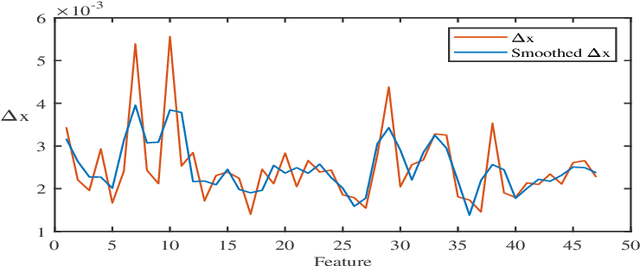
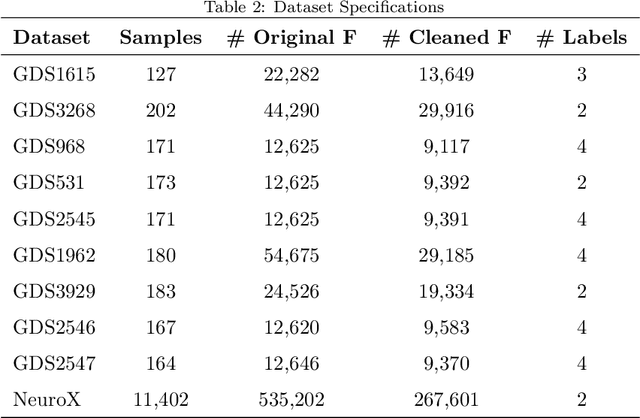
In the presence of large dimensional datasets that contain many irrelevant features (variables), dimensionality reduction algorithms have proven to be useful in removing features with low variance and combine features with high correlation. In this paper, we propose a new feature selection method which uses singular value decomposition of a matrix and the method of least squares to remove the irrelevant features and detect correlations between the remaining features. The effectiveness of our method has been verified by performing a series of comparisons with state-of-the-art feature selection methods over ten genetic datasets ranging up from 9,117 to 267,604 features. The results show that our method is favorable in various aspects compared to state-of-the-art feature selection methods.
A Feature Selection Based on Perturbation Theory
Feb 26, 2019
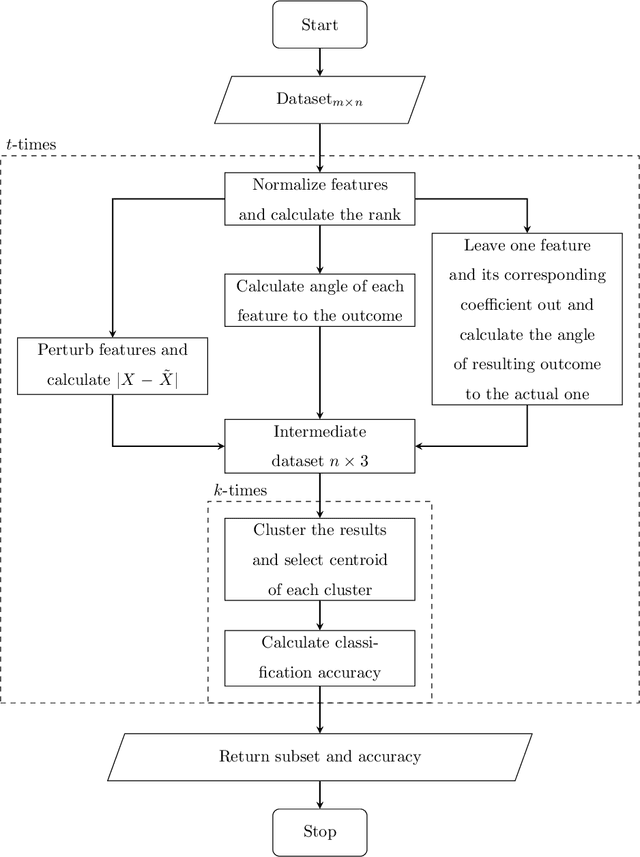


Consider a supervised dataset $D=[A\mid \textbf{b}]$, where $\textbf{b}$ is the outcome column, rows of $D$ correspond to observations, and columns of $A$ are the features of the dataset. A central problem in machine learning and pattern recognition is to select the most important features from $D$ to be able to predict the outcome. In this paper, we provide a new feature selection method where we use perturbation theory to detect correlations between features. We solve $AX=\textbf{b}$ using the method of least squares and singular value decomposition of $A$. In practical applications, such as in bioinformatics, the number of rows of $A$ (observations) are much less than the number of columns of $A$ (features). So we are dealing with singular matrices with big condition numbers. Although it is known that the solutions of least square problems in singular case are very sensitive to perturbations in $A$, our novel approach in this paper is to prove that the correlations between features can be detected by applying perturbations to $A$. The effectiveness of our method is verified by performing a series of comparisons with conventional and novel feature selection methods in the literature. It is demonstrated that in most situations, our method chooses considerably less number of features while attaining or exceeding the accuracy of the other methods.