 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFinetuning-Free Diffusion Model with Adaptive Constraint Guidance for Inorganic Crystal Structure Generation
Apr 14, 2026The discovery of inorganic crystal structures with targeted properties is a significant challenge in materials science. Generative models, especially state-of-the-art diffusion models, offer the promise of modeling complex data distributions and proposing novel, realistic samples. However, current generative AI models still struggle to produce diverse, original, and reliable structures of experimentally achievable materials suitable for high-stakes applications. In this work, we propose a generative machine learning framework based on diffusion models with adaptive constraint guidance, which enables the incorporation of user-defined physical and chemical constraints during the generation process. This approach is designed to be practical and interpretable for human experts, allowing transparent decision-making and expert-driven exploration. To ensure the robustness and validity of the generated candidates, we introduce a multi-step validation pipeline that combines graph neural network estimators trained to achieve DFT-level accuracy and convex hull analysis for assessing thermodynamic stability. Our approach has been tested and validated on several classical examples of inorganic families of compounds, as case studies. As a consequence, these preliminary results demonstrate our framework's ability to generate thermodynamically plausible crystal structures that satisfy targeted geometric constraints across diverse inorganic chemical systems.
Quantum-Aware Generative AI for Materials Discovery: A Framework for Robust Exploration Beyond DFT Biases
Dec 13, 2025Conventional generative models for materials discovery are predominantly trained and validated using data from Density Functional Theory (DFT) with approximate exchange-correlation functionals. This creates a fundamental bottleneck: these models inherit DFT's systematic failures for strongly correlated systems, leading to exploration biases and an inability to discover materials where DFT predictions are qualitatively incorrect. We introduce a quantum-aware generative AI framework that systematically addresses this limitation through tight integration of multi-fidelity learning and active validation. Our approach employs a diffusion-based generator conditioned on quantum-mechanical descriptors and a validator using an equivariant neural network potential trained on a hierarchical dataset spanning multiple levels of theory (PBE, SCAN, HSE06, CCSD(T)). Crucially, we implement a robust active learning loop that quantifies and targets the divergence between low- and high-fidelity predictions. We conduct comprehensive ablation studies to deconstruct the contribution of each component, perform detailed failure mode analysis, and benchmark our framework against state-of-the-art generative models (CDVAE, GNoME, DiffCSP) across several challenging material classes. Our results demonstrate significant practical gains: a 3-5x improvement in successfully identifying potentially stable candidates in high-divergence regions (e.g., correlated oxides) compared to DFT-only baselines, while maintaining computational feasibility. This work provides a rigorous, transparent framework for extending the effective search space of computational materials discovery beyond the limitations of single-fidelity models.
Mining experimental data from Materials Science literature with Large Language Models
Jan 19, 2024This study is dedicated to evaluating the capabilities of advanced large language models (LLMs) such as GPT-3.5-Turbo, GPT-4, and GPT-4-Turbo in the extraction of structured information from scientific documents within the field of materials science. We introduce a novel methodology for the comparative analysis of intricate material expressions, emphasising the standardisation of chemical formulas to tackle the complexities inherent in materials science information assessment. To this end, we primarily focus on two critical tasks of information extraction: (i) a named entity recognition (NER) of studied materials and physical properties and (ii) a relation extraction (RE) between these entities. The performance of LLMs in executing these tasks is benchmarked against traditional models based on the BERT architecture and rule-based approaches. For NER, LLMs fail to outperform the baseline with zero-shot prompting and exhibit only limited improvement with few-shot prompting. However, for RE, a GPT-3.5-Turbo fine-tuned with the appropriate strategy outperforms all models, including the baseline. Without any fine-tuning, GPT-4 and GPT-4-Turbo display remarkable reasoning and relationship extraction capabilities after being provided with merely a couple of examples, surpassing the baseline. Overall, the results suggest that although LLMs demonstrate relevant reasoning skills in connecting concepts, for tasks requiring extracting complex domain-specific entities like materials, specialised models are currently a better choice.
SMILES-X: autonomous molecular compounds characterization for small datasets without descriptors
Jul 04, 2019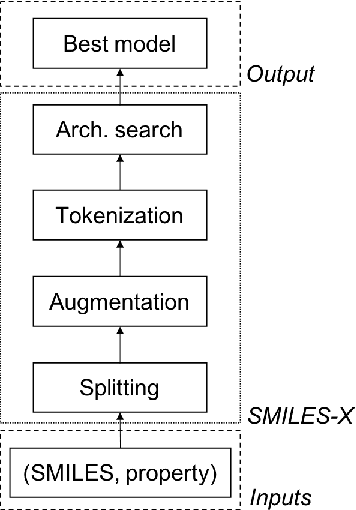

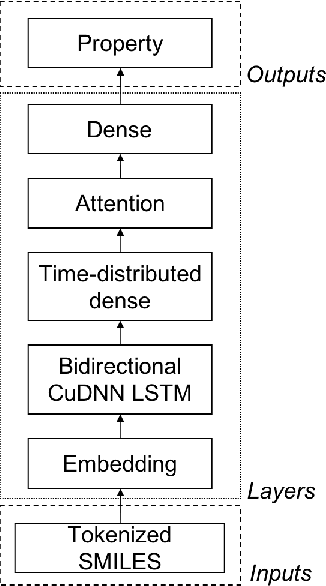
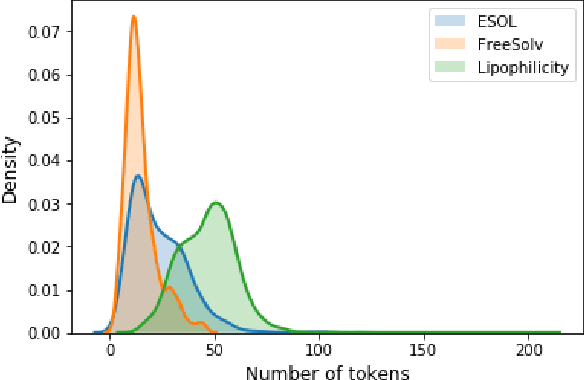
There is more and more evidence that machine learning can be successfully applied in materials science and related fields. However, datasets in these fields are often quite small ($\ll1000$ samples). It makes the most advanced machine learning techniques remain neglected, as they are considered to be applicable to big data only. Moreover, materials informatics methods often rely on human-engineered descriptors, that should be carefully chosen, or even created, to fit the physicochemical property that one intends to predict. In this article, we propose a new method that tackles both the issue of small datasets and the difficulty of task-specific descriptors development. The SMILES-X is an autonomous pipeline for molecular compounds characterisation based on a \{Embed-Encode-Attend-Predict\} neural architecture with a data-specific Bayesian hyper-parameters optimisation. The only input to the architecture -- the SMILES strings -- are de-canonicalised in order to efficiently augment the data. One of the key features of the architecture is the attention mechanism, which enables the interpretation of output predictions without extra computational cost. The SMILES-X shows new state-of-the-art results in the inference of aqueous solubility ($\overline{RMSE}_{test} \simeq 0.57 \pm 0.07$ mols/L), hydration free energy ($\overline{RMSE}_{test} \simeq 0.81 \pm 0.22$ kcal/mol, which is $\sim 24.5\%$ better than molecular dynamics simulations), and octanol/water distribution coefficient ($\overline{RMSE}_{test} \simeq 0.59 \pm 0.02$ for LogD at pH 7.4) of molecular compounds. The SMILES-X is intended to become an important asset in the toolkit of materials scientists and chemists. The source code for the SMILES-X is available at \href{https://github.com/GLambard/SMILES-X}{github.com/GLambard/SMILES-X}.