 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSMILES-X: autonomous molecular compounds characterization for small datasets without descriptors
Paper and Code
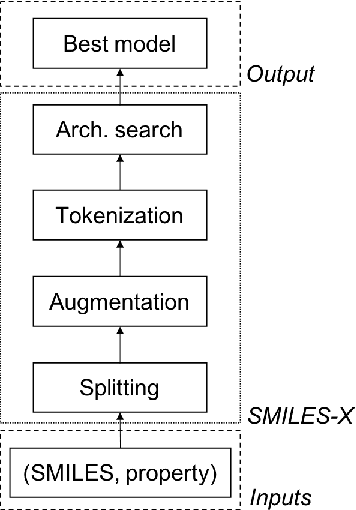

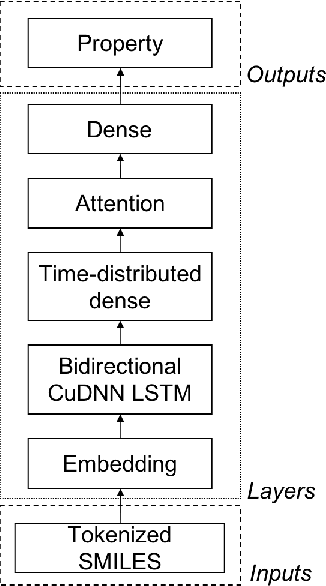
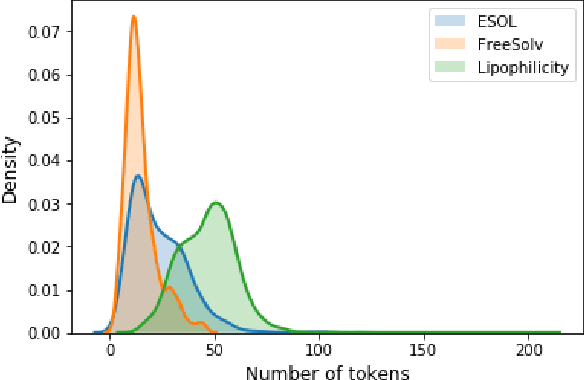
There is more and more evidence that machine learning can be successfully applied in materials science and related fields. However, datasets in these fields are often quite small ($\ll1000$ samples). It makes the most advanced machine learning techniques remain neglected, as they are considered to be applicable to big data only. Moreover, materials informatics methods often rely on human-engineered descriptors, that should be carefully chosen, or even created, to fit the physicochemical property that one intends to predict. In this article, we propose a new method that tackles both the issue of small datasets and the difficulty of task-specific descriptors development. The SMILES-X is an autonomous pipeline for molecular compounds characterisation based on a \{Embed-Encode-Attend-Predict\} neural architecture with a data-specific Bayesian hyper-parameters optimisation. The only input to the architecture -- the SMILES strings -- are de-canonicalised in order to efficiently augment the data. One of the key features of the architecture is the attention mechanism, which enables the interpretation of output predictions without extra computational cost. The SMILES-X shows new state-of-the-art results in the inference of aqueous solubility ($\overline{RMSE}_{test} \simeq 0.57 \pm 0.07$ mols/L), hydration free energy ($\overline{RMSE}_{test} \simeq 0.81 \pm 0.22$ kcal/mol, which is $\sim 24.5\%$ better than molecular dynamics simulations), and octanol/water distribution coefficient ($\overline{RMSE}_{test} \simeq 0.59 \pm 0.02$ for LogD at pH 7.4) of molecular compounds. The SMILES-X is intended to become an important asset in the toolkit of materials scientists and chemists. The source code for the SMILES-X is available at \href{https://github.com/GLambard/SMILES-X}{github.com/GLambard/SMILES-X}.