 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeISLAND: In-Silico Prediction of Proteins Binding Affinity Using Sequence Descriptors
Mar 22, 2018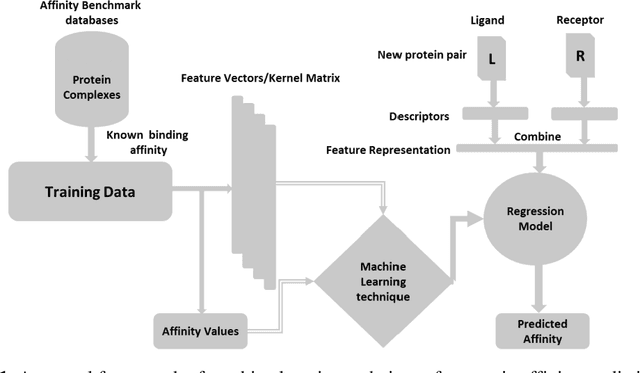
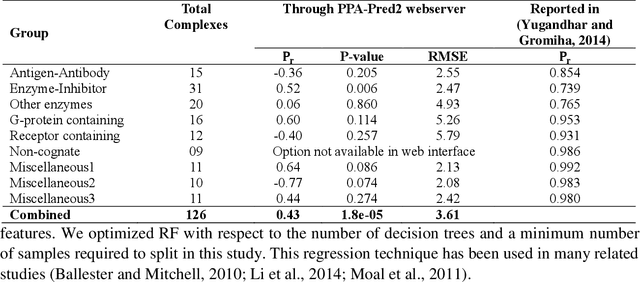
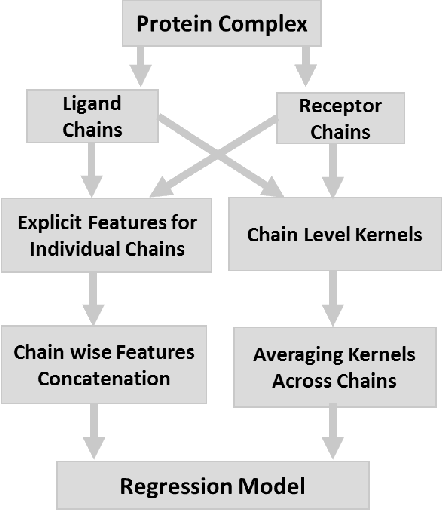
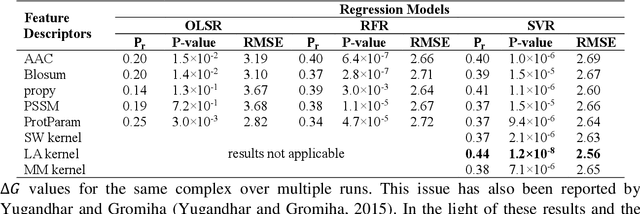
Determination of binding affinity of proteins in the formation of protein complexes requires sophisticated, expensive and time-consuming experimentation which can be replaced with computational methods. Most computational prediction techniques require protein structures which limit their applicability to protein complexes with known structures. In this work, we explore sequence based protein binding affinity prediction using machine learning. Our paper highlights the fact that the generalization performance of even the state of the art sequence-only predictor of binding affinity is far from satisfactory and that the development of effective and practical methods in this domain is still an open problem. We also propose a novel sequence-only predictor of binding affinity called ISLAND which gives better accuracy than existing methods over the same validation set as well as on external independent test dataset. A cloud-based webserver implementation of ISLAND and its Python code are available at the URL: http://faculty.pieas.edu.pk/fayyaz/software.html#island.
Training large margin host-pathogen protein-protein interaction predictors
Nov 21, 2017
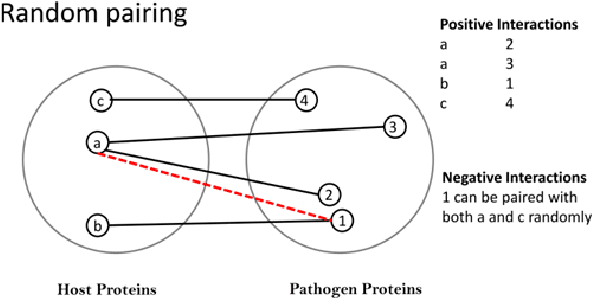
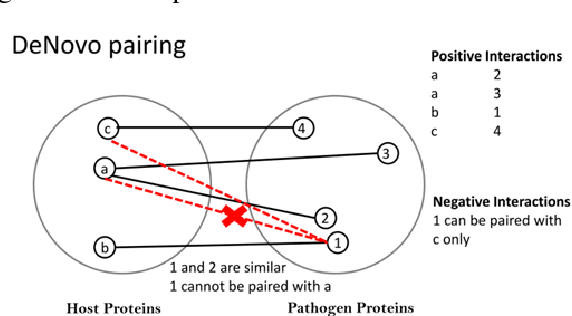
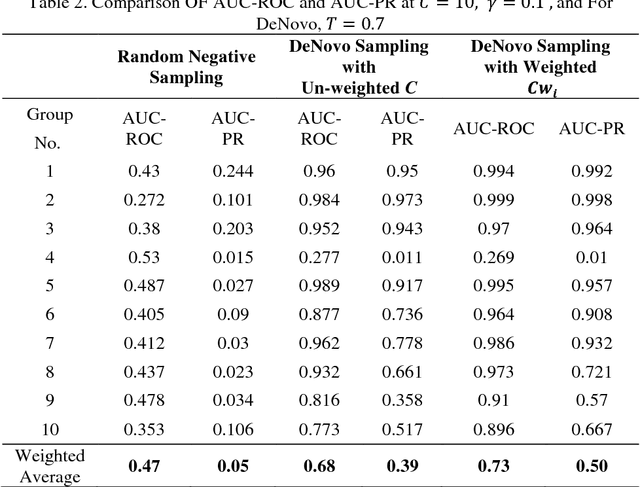
Detection of protein-protein interactions (PPIs) plays a vital role in molecular biology. Particularly, infections are caused by the interactions of host and pathogen proteins. It is important to identify host-pathogen interactions (HPIs) to discover new drugs to counter infectious diseases. Conventional wet lab PPI prediction techniques have limitations in terms of large scale application and budget. Hence, computational approaches are developed to predict PPIs. This study aims to develop large margin machine learning models to predict interspecies PPIs with a special interest in host-pathogen protein interactions (HPIs). Especially, we focus on seeking answers to three queries that arise while developing an HPI predictor. 1) How should we select negative samples? 2) What should be the size of negative samples as compared to the positive samples? 3) What type of margin violation penalty should be used to train the predictor? We compare two available methods for negative sampling. Moreover, we propose a new method of assigning weights to each training example in weighted SVM depending on the distance of the negative examples from the positive examples. We have also developed a web server for our HPI predictor called HoPItor (Host Pathogen Interaction predicTOR) that can predict interactions between human and viral proteins. This webserver can be accessed at the URL: http://faculty.pieas.edu.pk/fayyaz/software.html#HoPItor.