 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDefining Reference Sequences for Nocardia Species by Similarity and Clustering Analyses of 16S rRNA Gene Sequence Data
Nov 29, 2023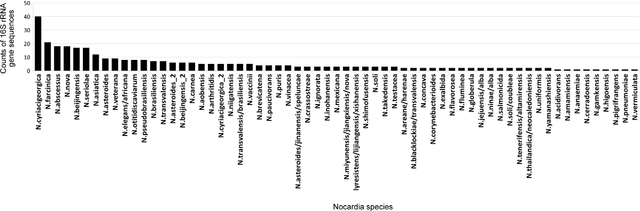
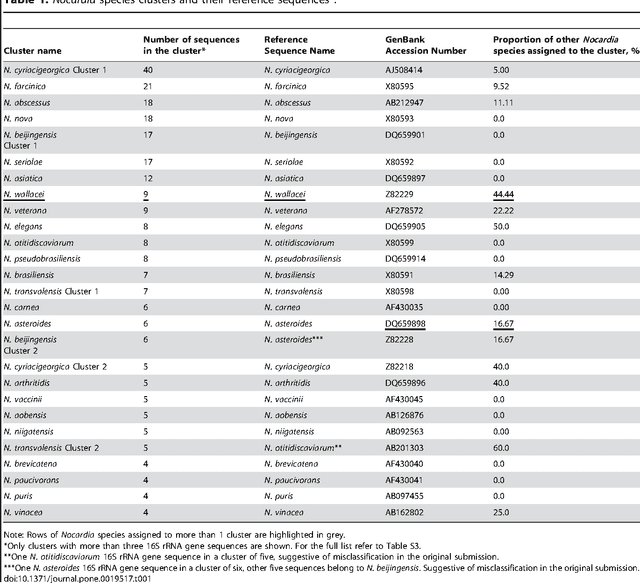
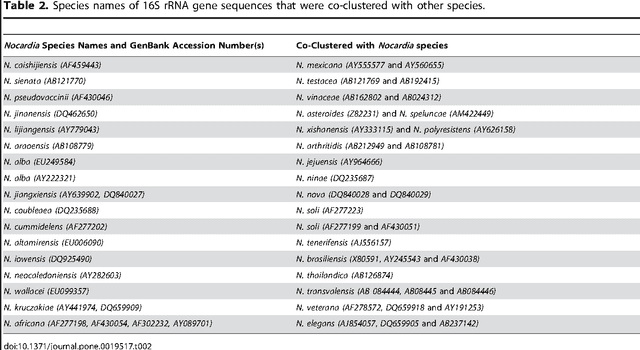
The intra- and inter-species genetic diversity of bacteria and the absence of 'reference', or the most representative, sequences of individual species present a significant challenge for sequence-based identification. The aims of this study were to determine the utility, and compare the performance of several clustering and classification algorithms to identify the species of 364 sequences of 16S rRNA gene with a defined species in GenBank, and 110 sequences of 16S rRNA gene with no defined species, all within the genus Nocardia. A total of 364 16S rRNA gene sequences of Nocardia species were studied. In addition, 110 16S rRNA gene sequences assigned only to the Nocardia genus level at the time of submission to GenBank were used for machine learning classification experiments. Different clustering algorithms were compared with a novel algorithm or the linear mapping (LM) of the distance matrix. Principal Components Analysis was used for the dimensionality reduction and visualization. Results: The LM algorithm achieved the highest performance and classified the set of 364 16S rRNA sequences into 80 clusters, the majority of which (83.52%) corresponded with the original species. The most representative 16S rRNA sequences for individual Nocardia species have been identified as 'centroids' in respective clusters from which the distances to all other sequences were minimized; 110 16S rRNA gene sequences with identifications recorded only at the genus level were classified using machine learning methods. Simple kNN machine learning demonstrated the highest performance and classified Nocardia species sequences with an accuracy of 92.7% and a mean frequency of 0.578.
Linear normalised hash function for clustering gene sequences and identifying reference sequences from multiple sequence alignments
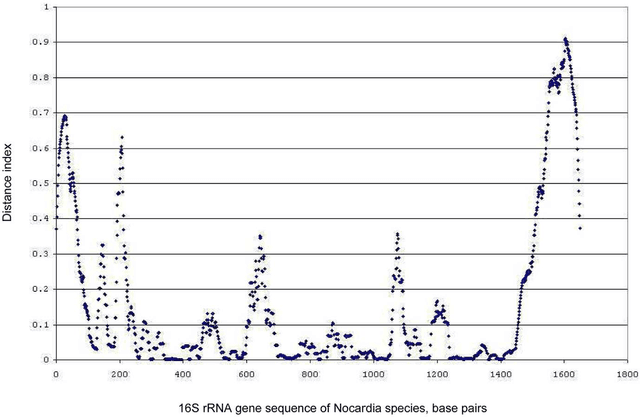
Nov 29, 2023The aim of this study was to develop a method that would identify the cluster centroids and the optimal number of clusters for a given sensitivity level and could work equally well for the different sequence datasets. A novel method that combines the linear mapping hash function and multiple sequence alignment (MSA) was developed. This method takes advantage of the already sorted by similarity sequences from the MSA output, and identifies the optimal number of clusters, clusters cut-offs, and clusters centroids that can represent reference gene vouchers for the different species. The linear mapping hash function can map an already ordered by similarity distance matrix to indices to reveal gaps in the values around which the optimal cut-offs of the different clusters can be identified. The method was evaluated using sets of closely related (16S rRNA gene sequences of Nocardia species) and highly variable (VP1 genomic region of Enterovirus 71) sequences and outperformed existing unsupervised machine learning clustering methods and dimensionality reduction methods. This method does not require prior knowledge of the number of clusters or the distance between clusters, handles clusters of different sizes and shapes, and scales linearly with the dataset. The combination of MSA with the linear mapping hash function is a computationally efficient way of gene sequence clustering and can be a valuable tool for the assessment of similarity, clustering of different microbial genomes, identifying reference sequences, and for the study of evolution of bacteria and viruses.