 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeComProScanner: A multi-agent based framework for composition-property structured data extraction from scientific literature
Oct 23, 2025Since the advent of various pre-trained large language models, extracting structured knowledge from scientific text has experienced a revolutionary change compared with traditional machine learning or natural language processing techniques. Despite these advances, accessible automated tools that allow users to construct, validate, and visualise datasets from scientific literature extraction remain scarce. We therefore developed ComProScanner, an autonomous multi-agent platform that facilitates the extraction, validation, classification, and visualisation of machine-readable chemical compositions and properties, integrated with synthesis data from journal articles for comprehensive database creation. We evaluated our framework using 100 journal articles against 10 different LLMs, including both open-source and proprietary models, to extract highly complex compositions associated with ceramic piezoelectric materials and corresponding piezoelectric strain coefficients (d33), motivated by the lack of a large dataset for such materials. DeepSeek-V3-0324 outperformed all models with a significant overall accuracy of 0.82. This framework provides a simple, user-friendly, readily-usable package for extracting highly complex experimental data buried in the literature to build machine learning or deep learning datasets.
Improving Omics-Based Classification: The Role of Feature Selection and Synthetic Data Generation
May 06, 2025Given the increasing complexity of omics datasets, a key challenge is not only improving classification performance but also enhancing the transparency and reliability of model decisions. Effective model performance and feature selection are fundamental for explainability and reliability. In many cases, high dimensional omics datasets suffer from limited number of samples due to clinical constraints, patient conditions, phenotypes rarity and others conditions. Current omics based classification models often suffer from narrow interpretability, making it difficult to discern meaningful insights where trust and reproducibility are critical. This study presents a machine learning based classification framework that integrates feature selection with data augmentation techniques to achieve high standard classification accuracy while ensuring better interpretability. Using the publicly available dataset (E MTAB 8026), we explore a bootstrap analysis in six binary classification scenarios to evaluate the proposed model's behaviour. We show that the proposed pipeline yields cross validated perfomance on small dataset that is conserved when the trained classifier is applied to a larger test set. Our findings emphasize the fundamental balance between accuracy and feature selection, highlighting the positive effect of introducing synthetic data for better generalization, even in scenarios with very limited samples availability.
Uncovering Population PK Covariates from VAE-Generated Latent Spaces
May 05, 2025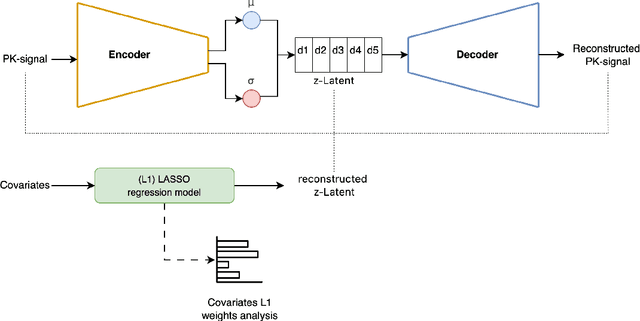
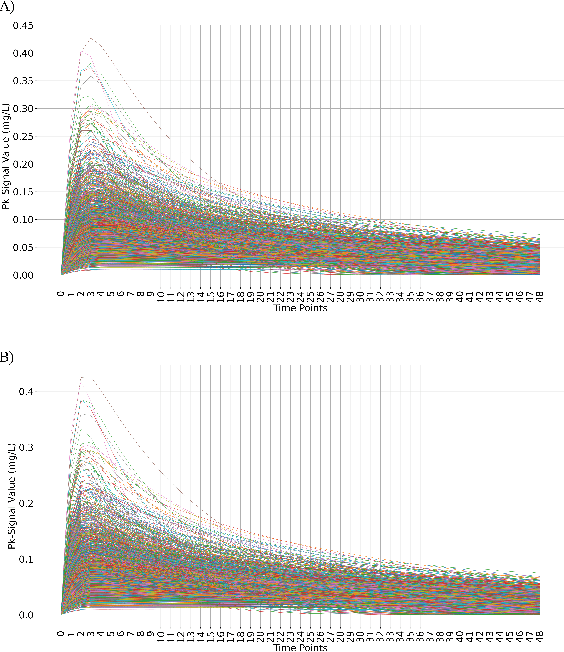
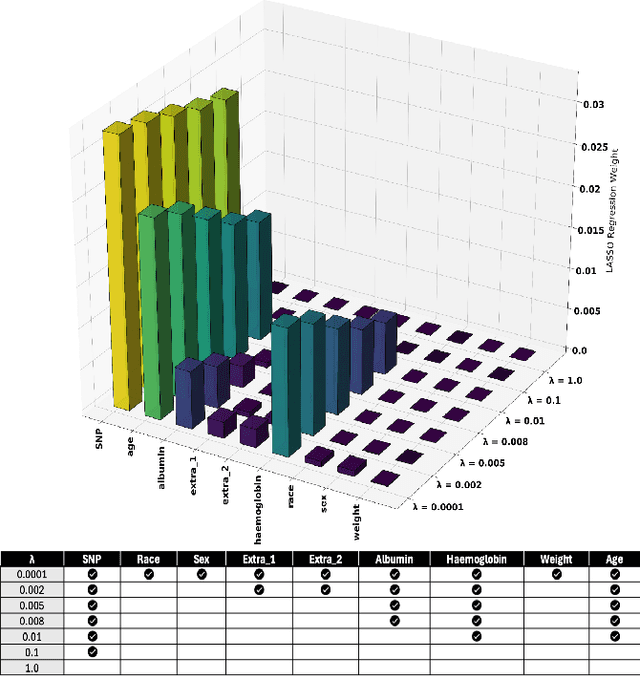

Population pharmacokinetic (PopPK) modelling is a fundamental tool for understanding drug behaviour across diverse patient populations and enabling personalized dosing strategies to improve therapeutic outcomes. A key challenge in PopPK analysis lies in identifying and modelling covariates that influence drug absorption, as these relationships are often complex and nonlinear. Traditional methods may fail to capture hidden patterns within the data. In this study, we propose a data-driven, model-free framework that integrates Variational Autoencoders (VAEs) deep learning model and LASSO regression to uncover key covariates from simulated tacrolimus pharmacokinetic (PK) profiles. The VAE compresses high-dimensional PK signals into a structured latent space, achieving accurate reconstruction with a mean absolute percentage error (MAPE) of 2.26%. LASSO regression is then applied to map patient-specific covariates to the latent space, enabling sparse feature selection through L1 regularization. This approach consistently identifies clinically relevant covariates for tacrolimus including SNP, age, albumin, and hemoglobin which are retained across the tested regularization strength levels, while effectively discarding non-informative features. The proposed VAE-LASSO methodology offers a scalable, interpretable, and fully data-driven solution for covariate selection, with promising applications in drug development and precision pharmacotherapy.
HistoSmith: Single-Stage Histology Image-Label Generation via Conditional Latent Diffusion for Enhanced Cell Segmentation and Classification
Feb 12, 2025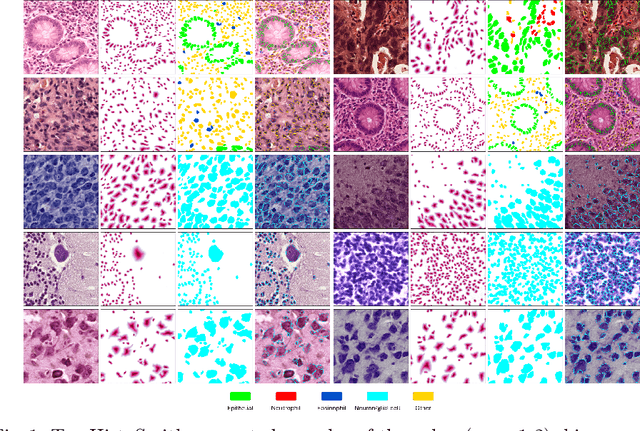
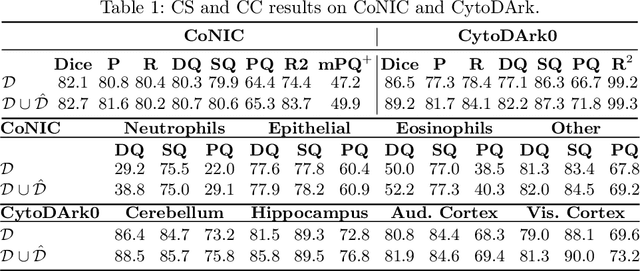
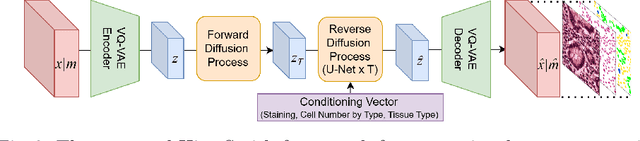
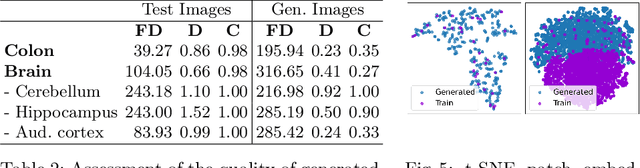
Precise segmentation and classification of cell instances are vital for analyzing the tissue microenvironment in histology images, supporting medical diagnosis, prognosis, treatment planning, and studies of brain cytoarchitecture. However, the creation of high-quality annotated datasets for training remains a major challenge. This study introduces a novel single-stage approach (HistoSmith) for generating image-label pairs to augment histology datasets. Unlike state-of-the-art methods that utilize diffusion models with separate components for label and image generation, our approach employs a latent diffusion model to learn the joint distribution of cellular layouts, classification masks, and histology images. This model enables tailored data generation by conditioning on user-defined parameters such as cell types, quantities, and tissue types. Trained on the Conic H&E histopathology dataset and the Nissl-stained CytoDArk0 dataset, the model generates realistic and diverse labeled samples. Experimental results demonstrate improvements in cell instance segmentation and classification, particularly for underrepresented cell types like neutrophils in the Conic dataset. These findings underscore the potential of our approach to address data scarcity challenges.
Mind the Gap: Evaluating Patch Embeddings from General-Purpose and Histopathology Foundation Models for Cell Segmentation and Classification
Feb 04, 2025Recent advancements in foundation models have transformed computer vision, driving significant performance improvements across diverse domains, including digital histopathology. However, the advantages of domain-specific histopathology foundation models over general-purpose models for specialized tasks such as cell analysis remain underexplored. This study investigates the representation learning gap between these two categories by analyzing multi-level patch embeddings applied to cell instance segmentation and classification. We implement an encoder-decoder architecture with a consistent decoder and various encoders. These include convolutional, vision transformer (ViT), and hybrid encoders pre-trained on ImageNet-22K or LVD-142M, representing general-purpose foundation models. These are compared against ViT encoders from the recently released UNI, Virchow2, and Prov-GigaPath foundation models, trained on patches extracted from hundreds of thousands of histopathology whole-slide images. The decoder integrates patch embeddings from different encoder depths via skip connections to generate semantic and distance maps. These maps are then post-processed to create instance segmentation masks where each label corresponds to an individual cell and to perform cell-type classification. All encoders remain frozen during training to assess their pre-trained feature extraction capabilities. Using the PanNuke and CoNIC histopathology datasets, and the newly introduced Nissl-stained CytoDArk0 dataset for brain cytoarchitecture studies, we evaluate instance-level detection, segmentation accuracy, and cell-type classification. This study provides insights into the comparative strengths and limitations of general-purpose vs. histopathology foundation models, offering guidance for model selection in cell-focused histopathology and brain cytoarchitecture analysis workflows.
Automated Classification of Cell Shapes: A Comparative Evaluation of Shape Descriptors
Nov 01, 2024
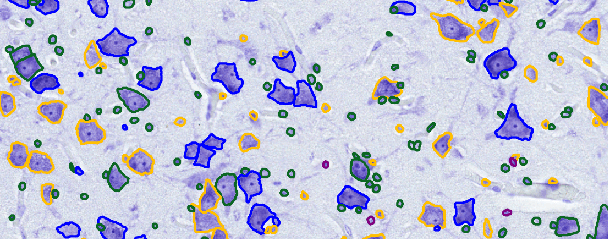
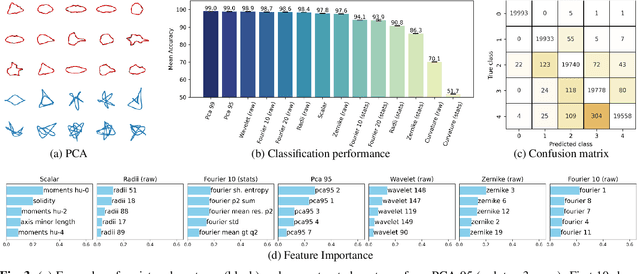
This study addresses the challenge of classifying cell shapes from noisy contours, such as those obtained through cell instance segmentation of histological images. We assess the performance of various features for shape classification, including Elliptical Fourier Descriptors, curvature features, and lower dimensional representations. Using an annotated synthetic dataset of noisy contours, we identify the most suitable shape descriptors and apply them to a set of real images for qualitative analysis. Our aim is to provide a comprehensive evaluation of descriptors for classifying cell shapes, which can support cell type identification and tissue characterization-critical tasks in both biological research and histopathological assessments.
CISCA and CytoDArk0: a Cell Instance Segmentation and Classification method for histo(patho)logical image Analyses and a new, open, Nissl-stained dataset for brain cytoarchitecture studies
Sep 06, 2024



Delineating and classifying individual cells in microscopy tissue images is a complex task, yet it is a pivotal endeavor in various medical and biological investigations. We propose a new deep learning framework (CISCA) for automatic cell instance segmentation and classification in histological slices to support detailed morphological and structural analysis or straightforward cell counting in digital pathology workflows and brain cytoarchitecture studies. At the core of CISCA lies a network architecture featuring a lightweight U-Net with three heads in the decoder. The first head classifies pixels into boundaries between neighboring cells, cell bodies, and background, while the second head regresses four distance maps along four directions. The network outputs from the first and second heads are integrated through a tailored post-processing step, which ultimately yields the segmentation of individual cells. A third head enables simultaneous classification of cells into relevant classes, if required. We showcase the effectiveness of our method using four datasets, including CoNIC, PanNuke, and MoNuSeg, which are publicly available H\&E datasets. Additionally, we introduce CytoDArk0, a novel dataset consisting of Nissl-stained images of the cortex, cerebellum, and hippocampus from mammals belonging to the orders Cetartiodactyla and Primates. We evaluate CISCA in comparison to other state-of-the-art methods, demonstrating CISCA's robustness and accuracy in segmenting and classifying cells across diverse tissue types, magnifications, and staining techniques.
Revealing Cortical Layers In Histological Brain Images With Self-Supervised Graph Convolutional Networks Applied To Cell-Graphs
Nov 26, 2023
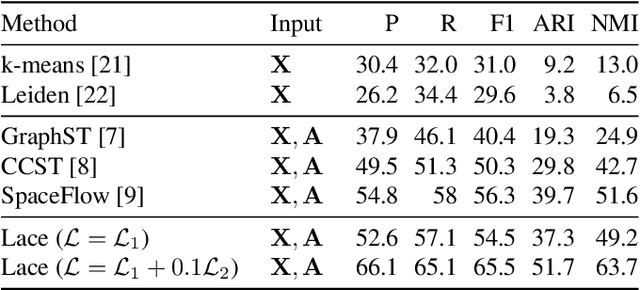

Identifying cerebral cortex layers is crucial for comparative studies of the cytoarchitecture aiming at providing insights into the relations between brain structure and function across species. The absence of extensive annotated datasets typically limits the adoption of machine learning approaches, leading to the manual delineation of cortical layers by neuroanatomists. We introduce a self-supervised approach to detect layers in 2D Nissl-stained histological slices of the cerebral cortex. It starts with the segmentation of individual cells and the creation of an attributed cell-graph. A self-supervised graph convolutional network generates cell embeddings that encode morphological and structural traits of the cellular environment and are exploited by a community detection algorithm for the final layering. Our method, the first self-supervised of its kind with no spatial transcriptomics data involved, holds the potential to accelerate cytoarchitecture analyses, sidestepping annotation needs and advancing cross-species investigation.
NCIS: Deep Color Gradient Maps Regression and Three-Class Pixel Classification for Enhanced Neuronal Cell Instance Segmentation in Nissl-Stained Histological Images
Jun 27, 2023



Deep learning has proven to be more effective than other methods in medical image analysis, including the seemingly simple but challenging task of segmenting individual cells, an essential step for many biological studies. Comparative neuroanatomy studies are an example where the instance segmentation of neuronal cells is crucial for cytoarchitecture characterization. This paper presents an end-to-end framework to automatically segment single neuronal cells in Nissl-stained histological images of the brain, thus aiming to enable solid morphological and structural analyses for the investigation of changes in the brain cytoarchitecture. A U-Net-like architecture with an EfficientNet as the encoder and two decoding branches is exploited to regress four color gradient maps and classify pixels into contours between touching cells, cell bodies, or background. The decoding branches are connected through attention gates to share relevant features, and their outputs are combined to return the instance segmentation of the cells. The method was tested on images of the cerebral cortex and cerebellum, outperforming other recent deep-learning-based approaches for the instance segmentation of cells.
MR-NOM: Multi-scale Resolution of Neuronal cells in Nissl-stained histological slices via deliberate Over-segmentation and Merging
Nov 14, 2022

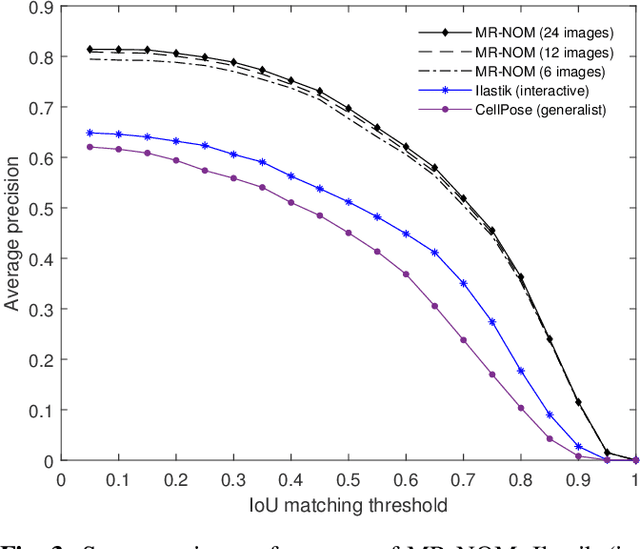
In comparative neuroanatomy, the characterization of brain cytoarchitecture is critical to a better understanding of brain structure and function, as it helps to distill information on the development, evolution, and distinctive features of different populations. The automatic segmentation of individual brain cells is a primary prerequisite and yet remains challenging. A new method (MR-NOM) was developed for the instance segmentation of cells in Nissl-stained histological images of the brain. MR-NOM exploits a multi-scale approach to deliberately over-segment the cells into superpixels and subsequently merge them via a classifier based on shape, structure, and intensity features. The method was tested on images of the cerebral cortex, proving successful in dealing with cells of varying characteristics that partially touch or overlap, showing better performance than two state-of-the-art methods.