 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA data-driven interpretation of the stability of molecular crystals
Sep 21, 2022
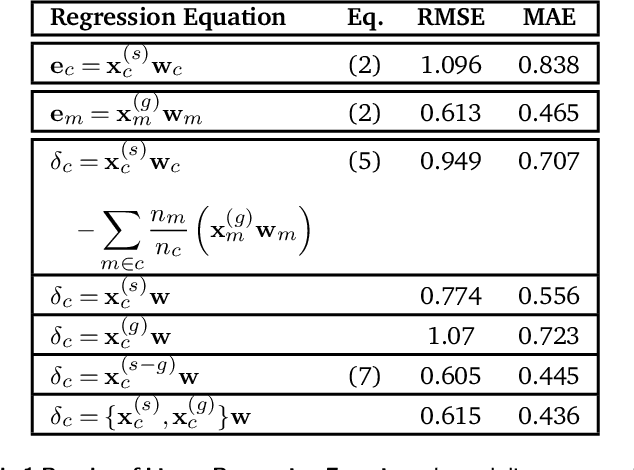
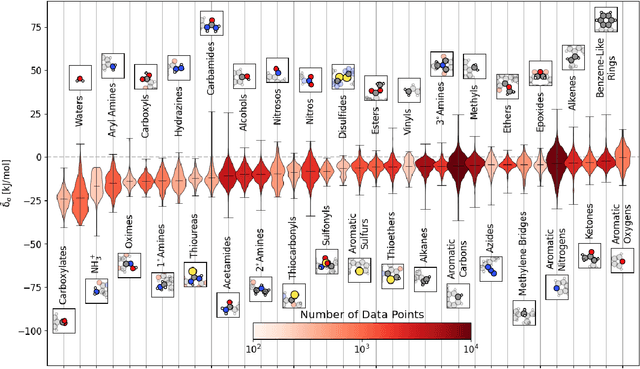
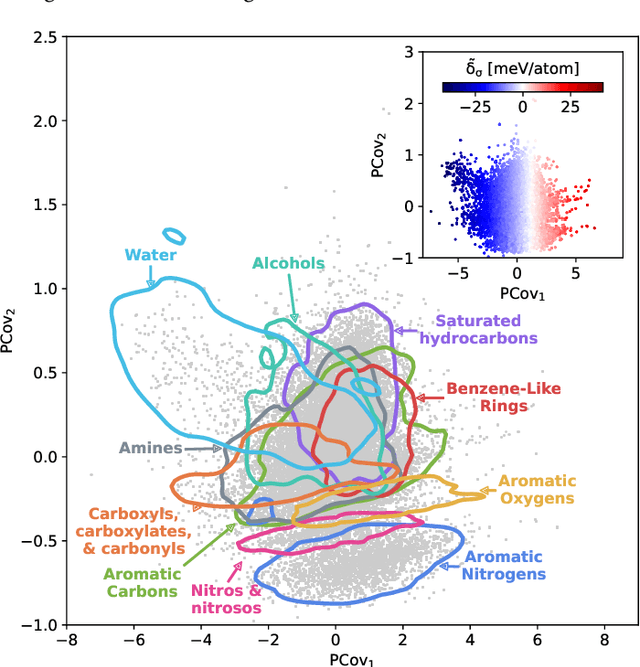
Due to the subtle balance of intermolecular interactions that govern structure-property relations, predicting the stability of crystal structures formed from molecular building blocks is a highly non-trivial scientific problem. A particularly active and fruitful approach involves classifying the different combinations of interacting chemical moieties, as understanding the relative energetics of different interactions enables the design of molecular crystals and fine-tuning their stabilities. While this is usually performed based on the empirical observation of the most commonly encountered motifs in known crystal structures, we propose to apply a combination of supervised and unsupervised machine-learning techniques to automate the construction of an extensive library of molecular building blocks. We introduce a structural descriptor tailored to the prediction of the binding energy for a curated dataset of organic crystals and exploit its atom-centered nature to obtain a data-driven assessment of the contribution of different chemical groups to the lattice energy of the crystal. We then interpret this library using a low-dimensional representation of the structure-energy landscape and discuss selected examples of the insights that can be extracted from this analysis, providing a complete database to guide the design of molecular materials.
Improving Sample and Feature Selection with Principal Covariates Regression
Dec 22, 2020

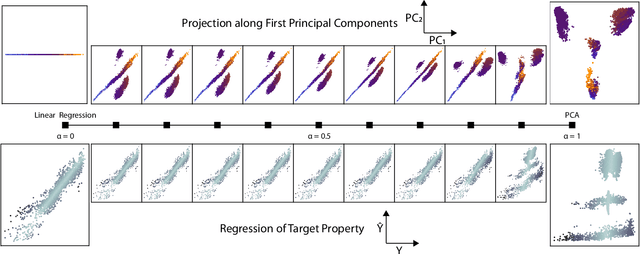
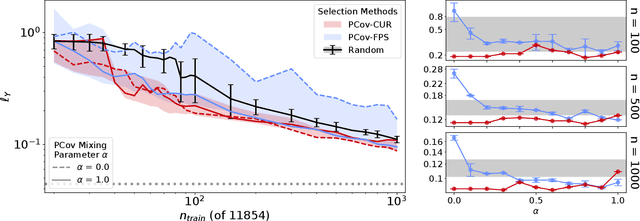
Selecting the most relevant features and samples out of a large set of candidates is a task that occurs very often in the context of automated data analysis, where it can be used to improve the computational performance, and also often the transferability, of a model. Here we focus on two popular sub-selection schemes which have been applied to this end: CUR decomposition, that is based on a low-rank approximation of the feature matrix and Farthest Point Sampling, that relies on the iterative identification of the most diverse samples and discriminating features. We modify these unsupervised approaches, incorporating a supervised component following the same spirit as the Principal Covariates Regression (PCovR) method. We show that incorporating target information provides selections that perform better in supervised tasks, which we demonstrate with ridge regression, kernel ridge regression, and sparse kernel regression. We also show that incorporating aspects of simple supervised learning models can improve the accuracy of more complex models, such as feed-forward neural networks. We present adjustments to minimize the impact that any subselection may incur when performing unsupervised tasks. We demonstrate the significant improvements associated with the use of PCov-CUR and PCov-FPS selections for applications to chemistry and materials science, typically reducing by a factor of two the number of features and samples which are required to achieve a given level of regression accuracy.