 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA data-driven interpretation of the stability of molecular crystals
Sep 21, 2022
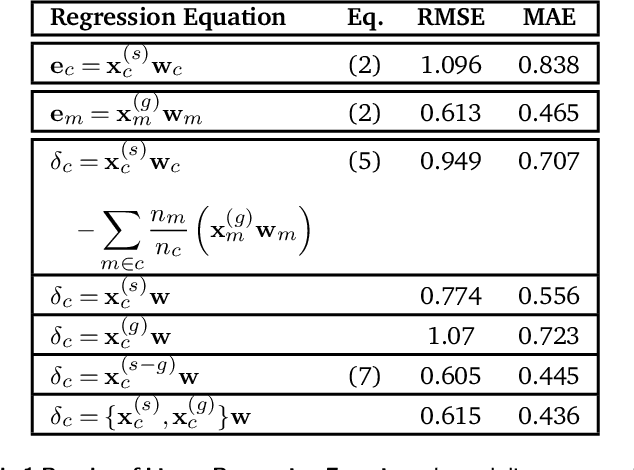
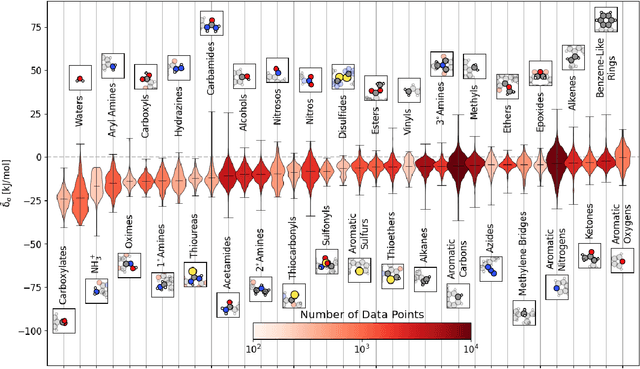
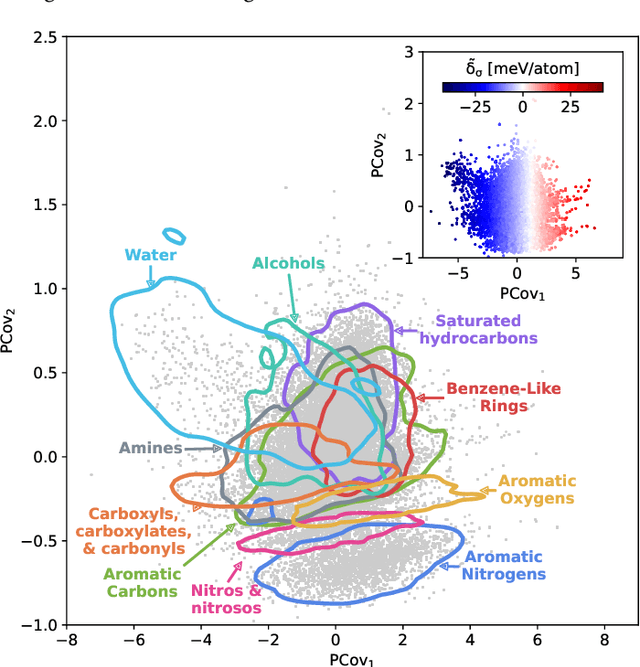
Due to the subtle balance of intermolecular interactions that govern structure-property relations, predicting the stability of crystal structures formed from molecular building blocks is a highly non-trivial scientific problem. A particularly active and fruitful approach involves classifying the different combinations of interacting chemical moieties, as understanding the relative energetics of different interactions enables the design of molecular crystals and fine-tuning their stabilities. While this is usually performed based on the empirical observation of the most commonly encountered motifs in known crystal structures, we propose to apply a combination of supervised and unsupervised machine-learning techniques to automate the construction of an extensive library of molecular building blocks. We introduce a structural descriptor tailored to the prediction of the binding energy for a curated dataset of organic crystals and exploit its atom-centered nature to obtain a data-driven assessment of the contribution of different chemical groups to the lattice energy of the crystal. We then interpret this library using a low-dimensional representation of the structure-energy landscape and discuss selected examples of the insights that can be extracted from this analysis, providing a complete database to guide the design of molecular materials.