 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBayesian group latent factor analysis with structured sparsity
Nov 11, 2015

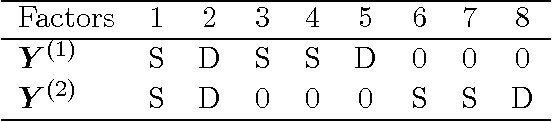

Latent factor models are the canonical statistical tool for exploratory analyses of low-dimensional linear structure for an observation matrix with p features across n samples. We develop a structured Bayesian group factor analysis model that extends the factor model to multiple coupled observation matrices; in the case of two observations, this reduces to a Bayesian model of canonical correlation analysis. The main contribution of this work is to carefully define a structured Bayesian prior that encourages both element-wise and column-wise shrinkage and leads to desirable behavior on high-dimensional data. In particular, our model puts a structured prior on the joint factor loading matrix, regularizing at three levels, which enables element-wise sparsity and unsupervised recovery of latent factors corresponding to structured variance across arbitrary subsets of the observations. In addition, our structured prior allows for both dense and sparse latent factors so that covariation among either all features or only a subset of features can both be recovered. We use fast parameter-expanded expectation-maximization for parameter estimation in this model. We validate our method on both simulated data with substantial structure and real data, comparing against a number of state-of-the-art approaches. These results illustrate useful properties of our model, including i) recovering sparse signal in the presence of dense effects; ii) the ability to scale naturally to large numbers of observations; iii) flexible observation- and factor-specific regularization to recover factors with a wide variety of sparsity levels and percentage of variance explained; and iv) tractable inference that scales to modern genomic and document data sizes.
Differential gene co-expression networks via Bayesian biclustering models
Nov 07, 2014



Identifying latent structure in large data matrices is essential for exploring biological processes. Here, we consider recovering gene co-expression networks from gene expression data, where each network encodes relationships between genes that are locally co-regulated by shared biological mechanisms. To do this, we develop a Bayesian statistical model for biclustering to infer subsets of co-regulated genes whose covariation may be observed in only a subset of the samples. Our biclustering method, BicMix, has desirable properties, including allowing overcomplete representations of the data, computational tractability, and jointly modeling unknown confounders and biological signals. Compared with related biclustering methods, BicMix recovers latent structure with higher precision across diverse simulation scenarios. Further, we develop a method to recover gene co-expression networks from the estimated sparse biclustering matrices. We apply BicMix to breast cancer gene expression data and recover a gene co-expression network that is differential across ER+ and ER- samples.