 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEDEN : An Event DEtection Network for the annotation of Breast Cancer recurrences in administrative claims data
Nov 15, 2022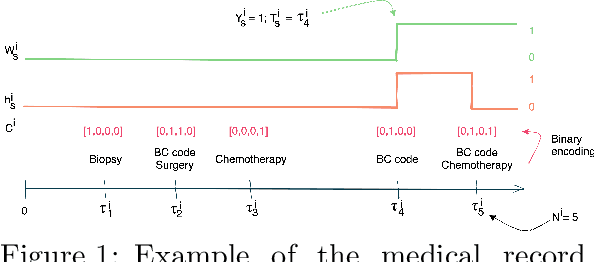



While the emergence of large administrative claims data provides opportunities for research, their use remains limited by the lack of clinical annotations relevant to disease outcomes, such as recurrence in breast cancer (BC). Several challenges arise from the annotation of such endpoints in administrative claims, including the need to infer both the occurrence and the date of the recurrence, the right-censoring of data, or the importance of time intervals between medical visits. Deep learning approaches have been successfully used to label temporal medical sequences, but no method is currently able to handle simultaneously right-censoring and visit temporality to detect survival events in medical sequences. We propose EDEN (Event DEtection Network), a time-aware Long-Short-Term-Memory network for survival analyses, and its custom loss function. Our method outperforms several state-of-the-art approaches on real-world BC datasets. EDEN constitutes a powerful tool to annotate disease recurrence from administrative claims, thus paving the way for the massive use of such data in BC research.
Network-Guided Biomarker Discovery
Dec 15, 2016
Identifying measurable genetic indicators (or biomarkers) of a specific condition of a biological system is a key element of precision medicine. Indeed it allows to tailor diagnostic, prognostic and treatment choice to individual characteristics of a patient. In machine learning terms, biomarker discovery can be framed as a feature selection problem on whole-genome data sets. However, classical feature selection methods are usually underpowered to process these data sets, which contain orders of magnitude more features than samples. This can be addressed by making the assumption that genetic features that are linked on a biological network are more likely to work jointly towards explaining the phenotype of interest. We review here three families of methods for feature selection that integrate prior knowledge in the form of networks.
Efficient network-guided multi-locus association mapping with graph cuts
Apr 18, 2013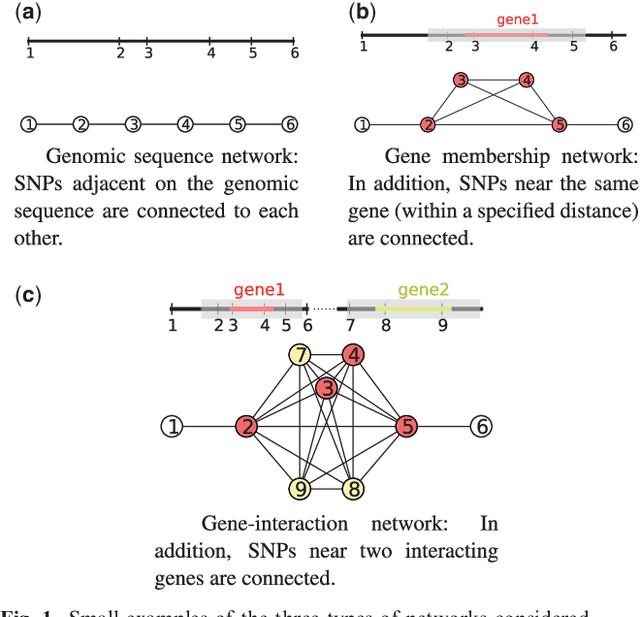
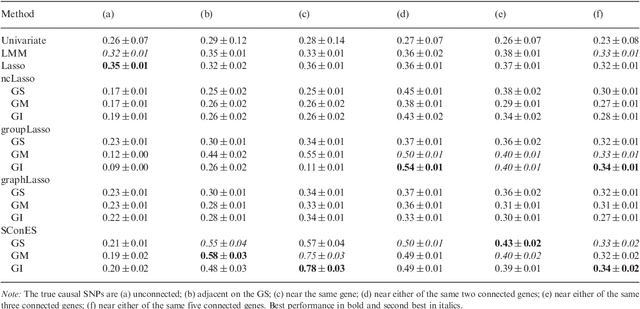
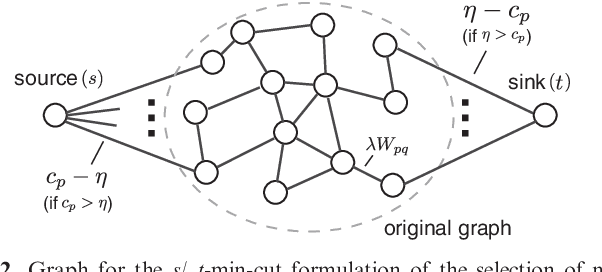

As an increasing number of genome-wide association studies reveal the limitations of attempting to explain phenotypic heritability by single genetic loci, there is growing interest for associating complex phenotypes with sets of genetic loci. While several methods for multi-locus mapping have been proposed, it is often unclear how to relate the detected loci to the growing knowledge about gene pathways and networks. The few methods that take biological pathways or networks into account are either restricted to investigating a limited number of predetermined sets of loci, or do not scale to genome-wide settings. We present SConES, a new efficient method to discover sets of genetic loci that are maximally associated with a phenotype, while being connected in an underlying network. Our approach is based on a minimum cut reformulation of the problem of selecting features under sparsity and connectivity constraints that can be solved exactly and rapidly. SConES outperforms state-of-the-art competitors in terms of runtime, scales to hundreds of thousands of genetic loci, and exhibits higher power in detecting causal SNPs in simulation studies than existing methods. On flowering time phenotypes and genotypes from Arabidopsis thaliana, SConES detects loci that enable accurate phenotype prediction and that are supported by the literature. Matlab code for SConES is available at http://webdav.tuebingen.mpg.de/u/karsten/Forschung/scones/