 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHierarchy exploitation to detect missing annotations on hierarchical multi-label classification
Jul 13, 2022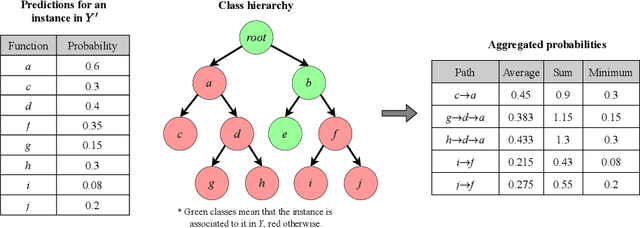
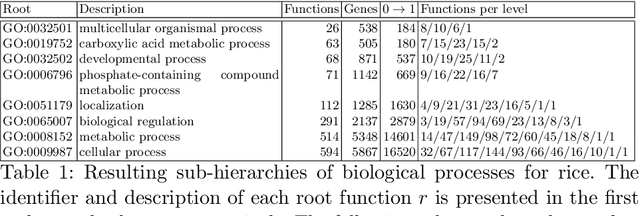
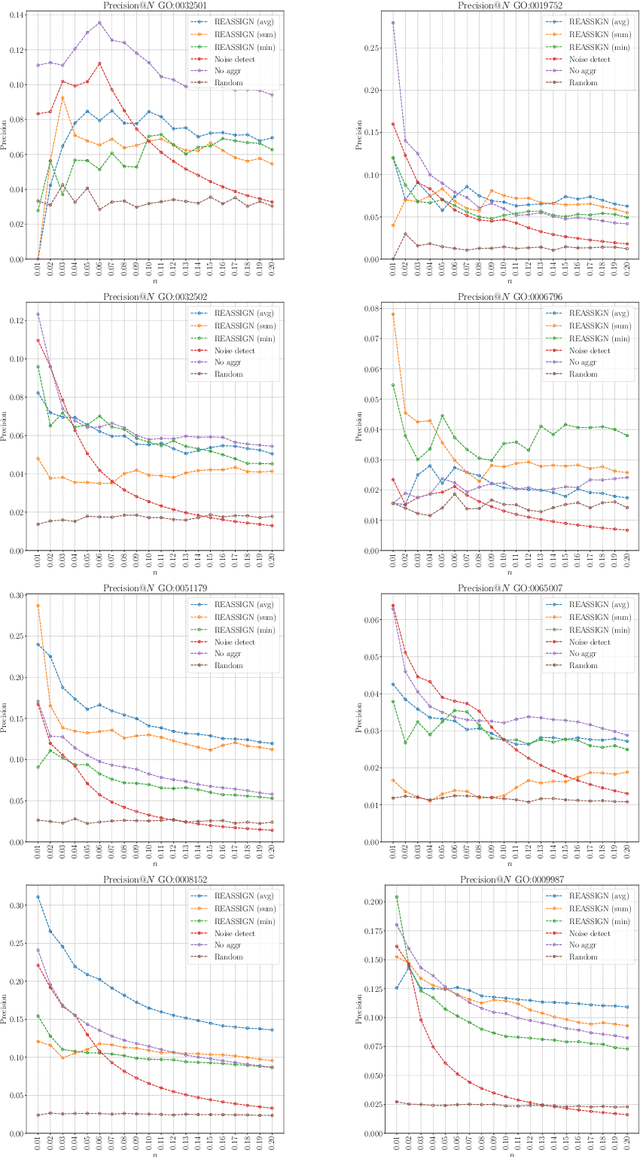
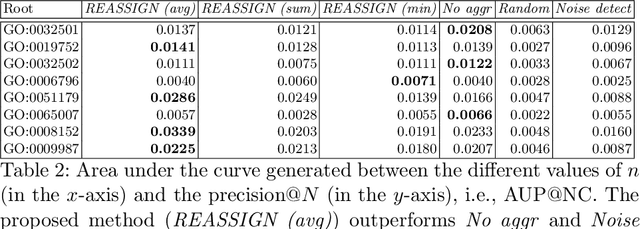
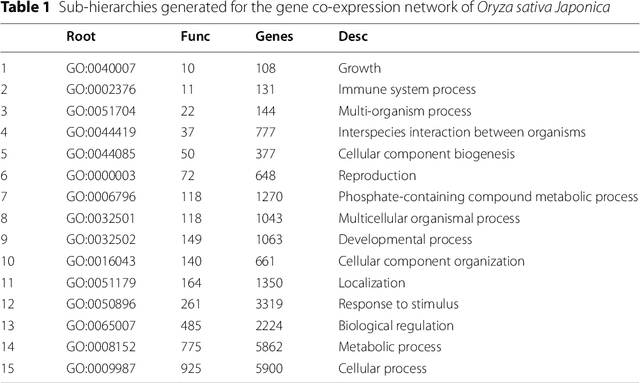
The availability of genomic data has grown exponentially in the last decade, mainly due to the development of new sequencing technologies. Based on the interactions between genes (and gene products) extracted from the increasing genomic data, numerous studies have focused on the identification of associations between genes and functions. While these studies have shown great promise, the problem of annotating genes with functions remains an open challenge. In this work, we present a method to detect missing annotations in hierarchical multi-label classification datasets. We propose a method that exploits the class hierarchy by computing aggregated probabilities to the paths of classes from the leaves to the root for each instance. The proposed method is presented in the context of predicting missing gene function annotations, where these aggregated probabilities are further used to select a set of annotations to be verified through in vivo experiments. The experiments on Oriza sativa Japonica, a variety of rice, showcase that incorporating the hierarchy of classes into the method often improves the predictive performance and our proposed method yields superior results when compared to competitor methods from the literature.
Feature extraction using Spectral Clustering for Gene Function Prediction
Mar 25, 2022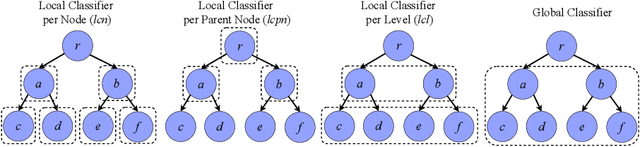

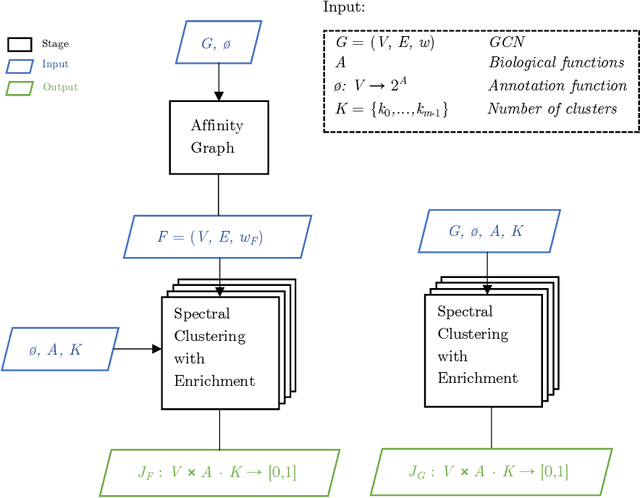

Gene annotation addresses the problem of predicting unknown associations between gene and functions (e.g., biological processes) of a specific organism. Despite recent advances, the cost and time demanded by annotation procedures that rely largely on in vivo biological experiments remain prohibitively high. This paper presents a novel in silico approach for to the annotation problem that combines cluster analysis and hierarchical multi-label classification (HMC). The approach uses spectral clustering to extract new features from the gene co-expression network (GCN) and enrich the prediction task. HMC is used to build multiple estimators that consider the hierarchical structure of gene functions. The proposed approach is applied to a case study on Zea mays, one of the most dominant and productive crops in the world. The results illustrate how in silico approaches are key to reduce the time and costs of gene annotation. More specifically, they highlight the importance of: (i) building new features that represent the structure of gene relationships in GCNs to annotate genes; and (ii) taking into account the structure of biological processes to obtain consistent predictions.
A Top-down Supervised Learning Approach to Hierarchical Multi-label Classification in Networks
Mar 23, 2022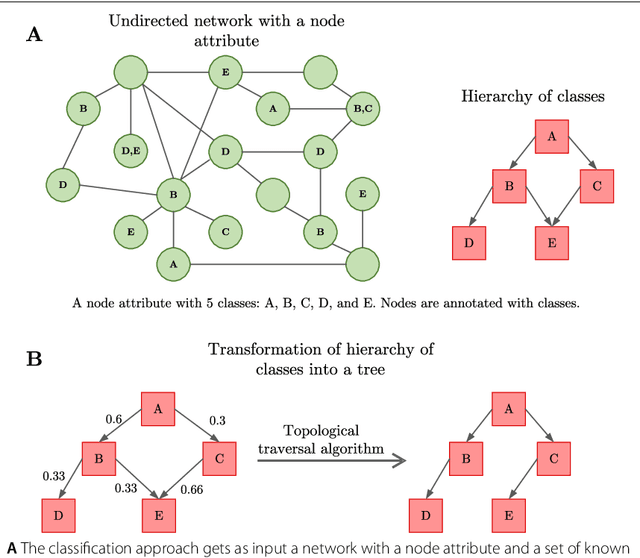
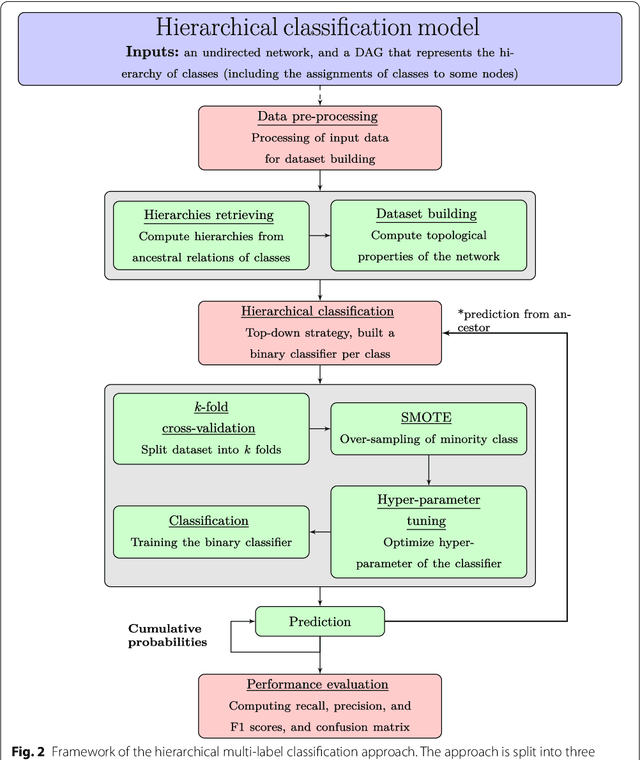

Node classification is the task of inferring or predicting missing node attributes from information available for other nodes in a network. This paper presents a general prediction model to hierarchical multi-label classification (HMC), where the attributes to be inferred can be specified as a strict poset. It is based on a top-down classification approach that addresses hierarchical multi-label classification with supervised learning by building a local classifier per class. The proposed model is showcased with a case study on the prediction of gene functions for Oryza sativa Japonica, a variety of rice. It is compared to the Hierarchical Binomial-Neighborhood, a probabilistic model, by evaluating both approaches in terms of prediction performance and computational cost. The results in this work support the working hypothesis that the proposed model can achieve good levels of prediction efficiency, while scaling up in relation to the state of the art.
Using Overlapping Communities and Network Structure for Identifying Reduced Groups of Stress Responsive Genes
Oct 23, 2020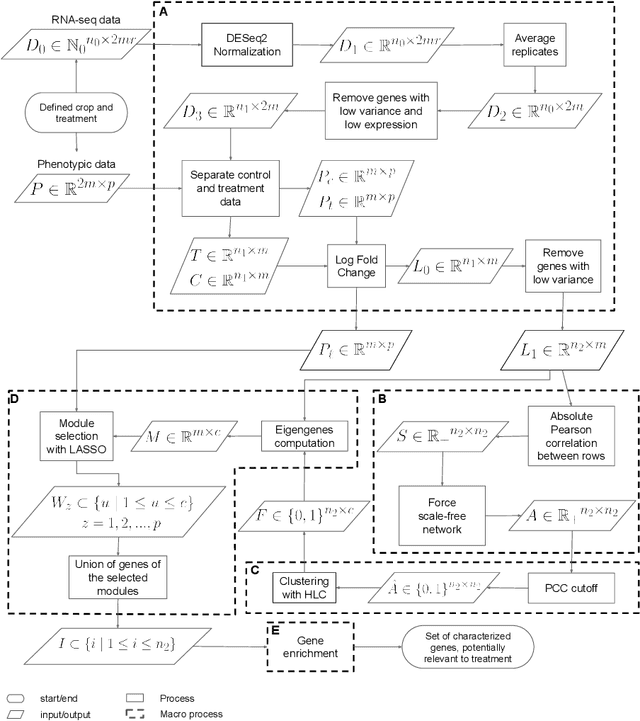


This paper proposes a workflow to identify genes responding to a specific treatment in an organism, such as abiotic stresses, a main cause of extensive agricultural production losses worldwide. On input RNA sequencing read counts (measured for genotypes under control and treatment conditions) and biological replicates, it outputs a collection of characterized genes, potentially relevant to treatment. Technically, the proposed approach is both a generalization and an extension of WGCNA; its main goal is to identify specific modules in a network of genes after a sequence of normalization and filtering steps. In this work, module detection is achieved by using Hierarchical Link Clustering, which can recognize overlapping communities and thus have more biological meaning given the overlapping regulatory domains of systems that generate co-expression. Additional steps and information are also added to the workflow, where some networks in the intermediate steps are forced to be scale-free and LASSO regression is employed to select the most significant modules of phenotypical responses to stress. Finally, the workflow is showcased with a systematic study on rice (Oryza sativa), a major food source that is known to be highly sensitive to salt stress: a total of 6 modules are detected as relevant in the response to salt stress in rice; these genes may act as potential targets for the improvement of salinity tolerance in rice cultivars. The proposed workflow has the potential to ultimately reduce the search-space for candidate genes responding to a specific treatment, which can considerably optimize the effort, time, and money invested by researchers in the experimental validation of stress responsive genes.
Spectral Evolution with Approximated Eigenvalue Trajectories for Link Prediction
Jun 22, 2020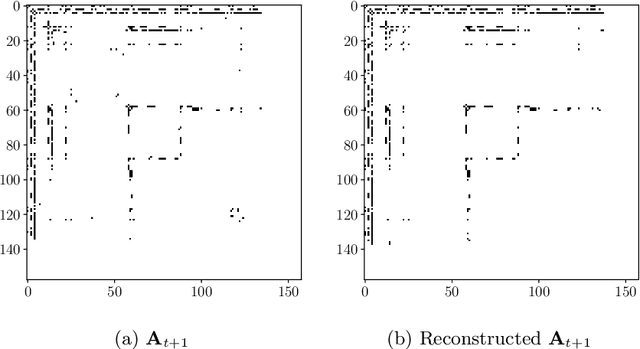
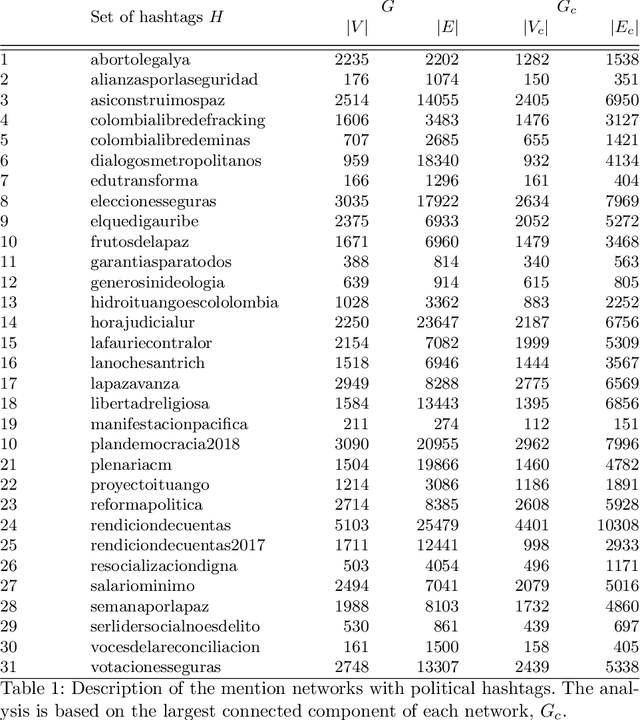


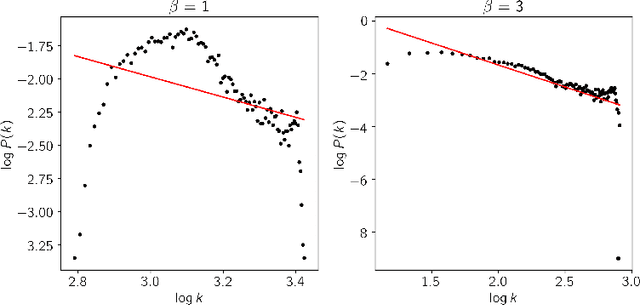
The spectral evolution model aims to characterize the growth of large networks (i.e., how they evolve as new edges are established) in terms of the eigenvalue decomposition of the adjacency matrices. It assumes that, while eigenvectors remain constant, eigenvalues evolve in a predictable manner over time. This paper extends the original formulation of the model twofold. First, it presents a method to compute an approximation of the spectral evolution of eigenvalues based on the Rayleigh quotient. Second, it proposes an algorithm to estimate the evolution of eigenvalues by extrapolating only a fraction of their approximated values. The proposed model is used to characterize mention networks of users who posted tweets that include the most popular political hashtags in Colombia from August 2017 to August 2018 (the period which concludes the disarmament of the Revolutionary Armed Forces of Colombia). To evaluate the extent to which the spectral evolution model resembles these networks, link prediction methods based on learning algorithms (i.e., extrapolation and regression) and graph kernels are implemented. Experimental results show that the learning algorithms deployed on the approximated trajectories outperform the usual kernel and extrapolation methods at predicting the formation of new edges.