 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUsing Overlapping Communities and Network Structure for Identifying Reduced Groups of Stress Responsive Genes
Oct 23, 2020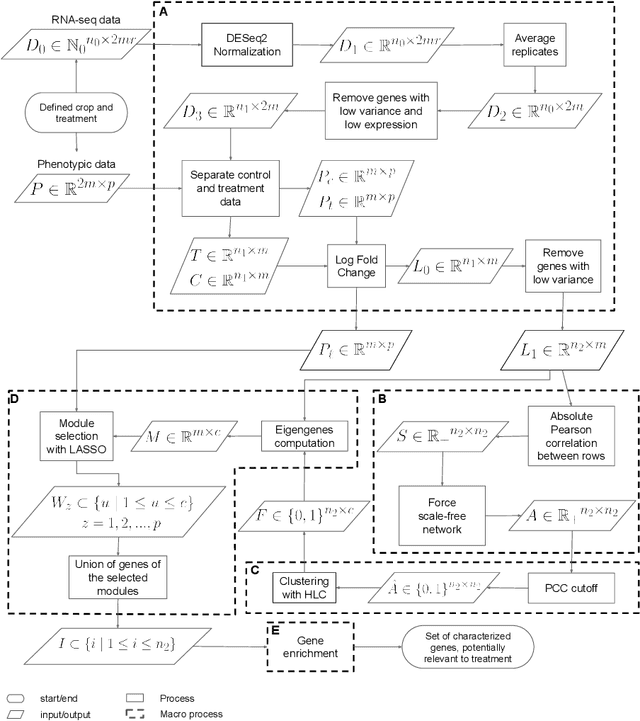
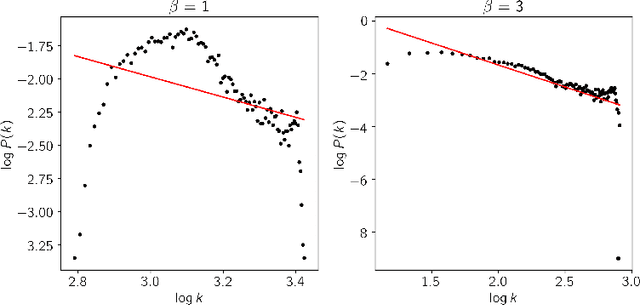


This paper proposes a workflow to identify genes responding to a specific treatment in an organism, such as abiotic stresses, a main cause of extensive agricultural production losses worldwide. On input RNA sequencing read counts (measured for genotypes under control and treatment conditions) and biological replicates, it outputs a collection of characterized genes, potentially relevant to treatment. Technically, the proposed approach is both a generalization and an extension of WGCNA; its main goal is to identify specific modules in a network of genes after a sequence of normalization and filtering steps. In this work, module detection is achieved by using Hierarchical Link Clustering, which can recognize overlapping communities and thus have more biological meaning given the overlapping regulatory domains of systems that generate co-expression. Additional steps and information are also added to the workflow, where some networks in the intermediate steps are forced to be scale-free and LASSO regression is employed to select the most significant modules of phenotypical responses to stress. Finally, the workflow is showcased with a systematic study on rice (Oryza sativa), a major food source that is known to be highly sensitive to salt stress: a total of 6 modules are detected as relevant in the response to salt stress in rice; these genes may act as potential targets for the improvement of salinity tolerance in rice cultivars. The proposed workflow has the potential to ultimately reduce the search-space for candidate genes responding to a specific treatment, which can considerably optimize the effort, time, and money invested by researchers in the experimental validation of stress responsive genes.