 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAtomistic structure search using local surrogate mode
Aug 19, 2022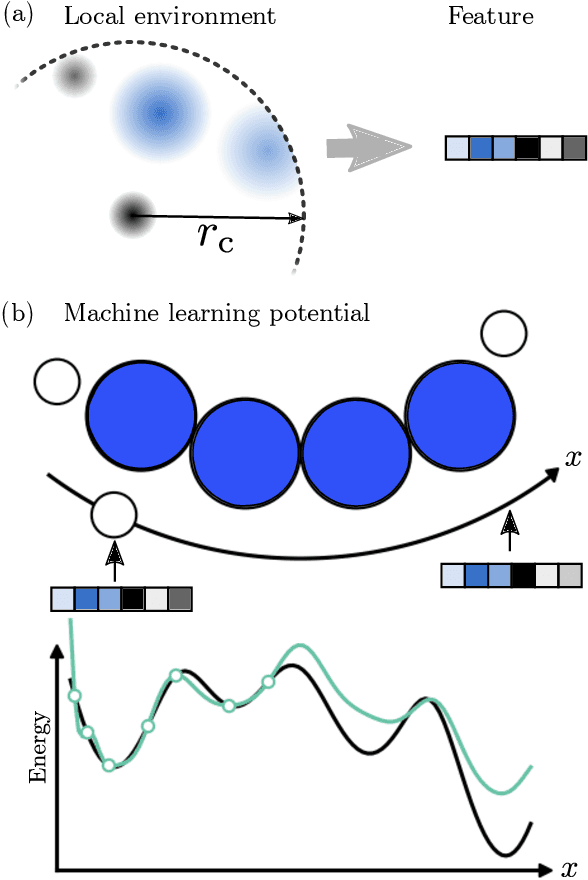
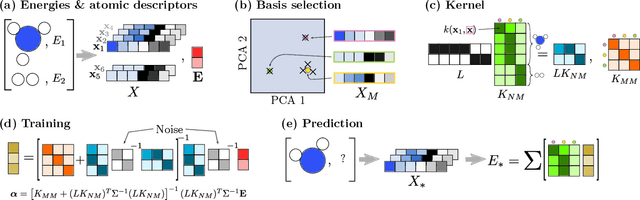
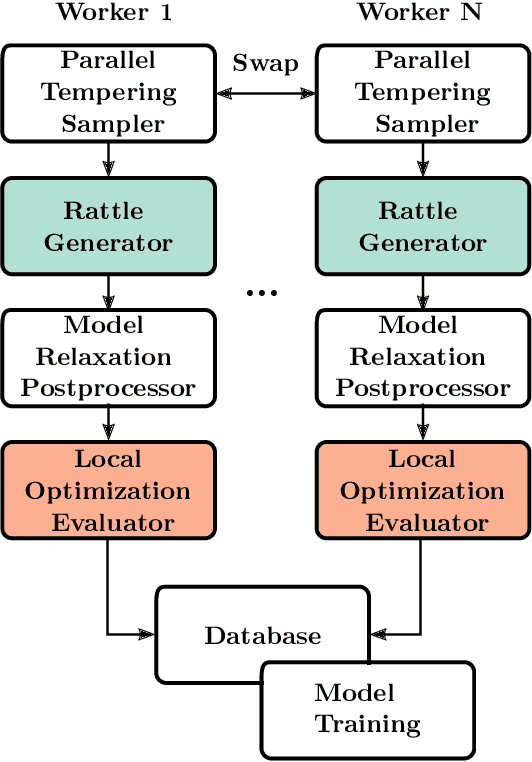

We describe a local surrogate model for use in conjunction with global structure search methods. The model follows the Gaussian approximation potential (GAP) formalism and is based on a the smooth overlap of atomic positions descriptor with sparsification in terms of a reduced number of local environments using mini-batch $k$-means. The model is implemented in the Atomistic Global Optimization X framework and used as a partial replacement of the local relaxations in basin hopping structure search. The approach is shown to be robust for a wide range of atomistic system including molecules, nano-particles, surface supported clusters and surface thin films. The benefits in a structure search context of a local surrogate model are demonstrated. This includes the ability to transfer learning from smaller systems as well as the possibility to perform concurrent multi-stoichiometry searches.
Generating stable molecules using imitation and reinforcement learning
Jul 11, 2021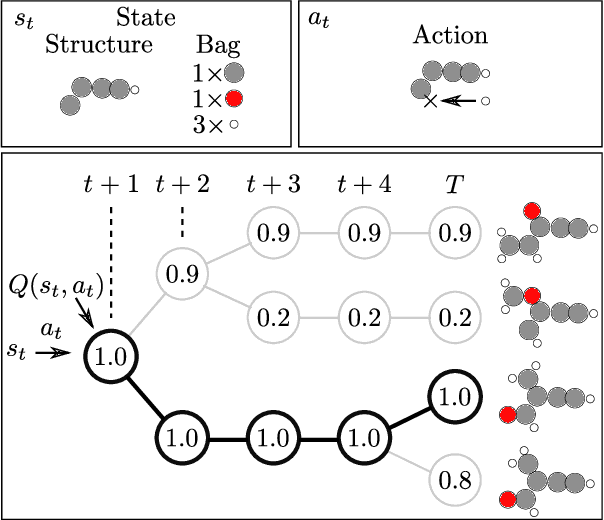
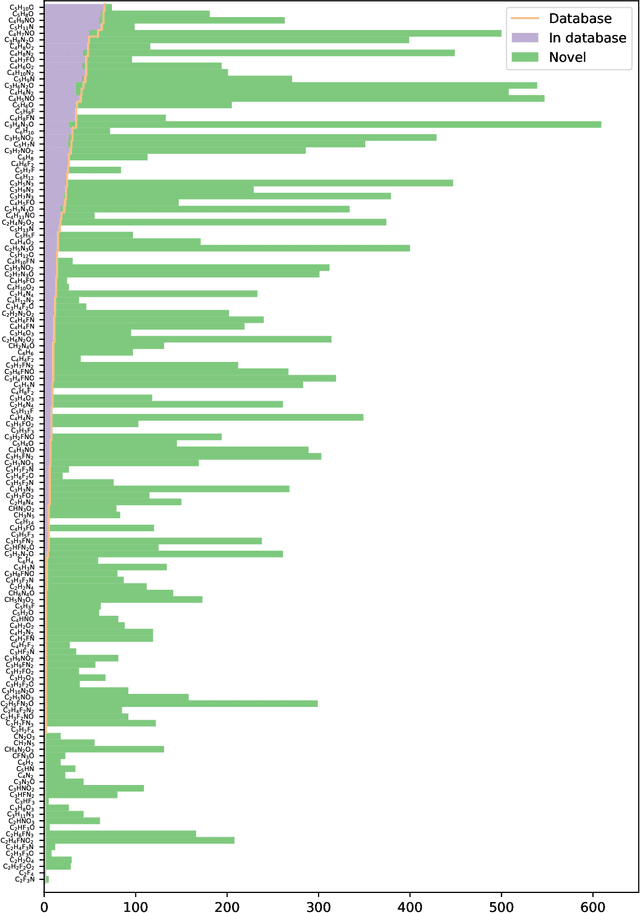
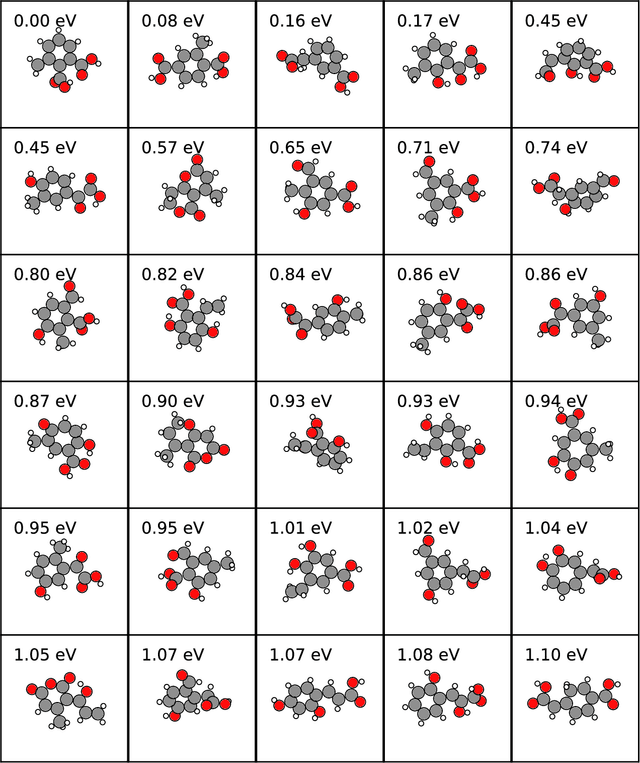
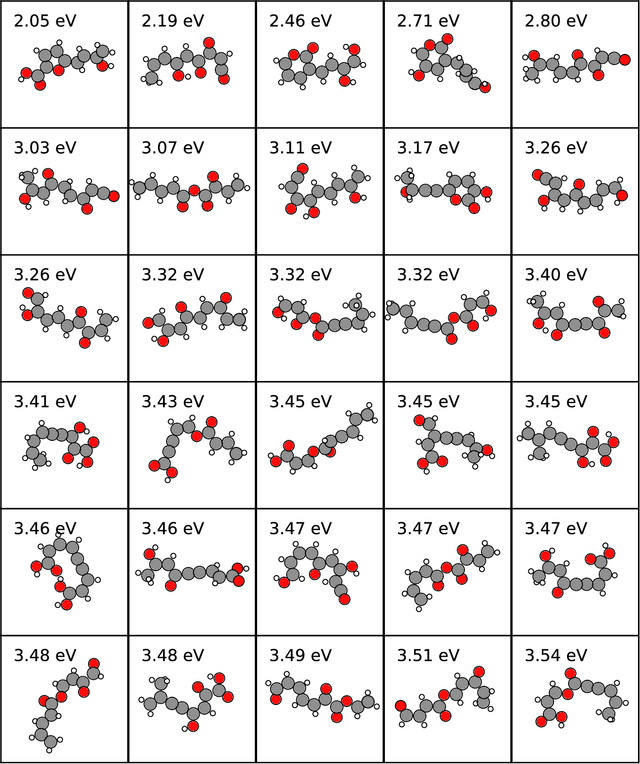
Chemical space is routinely explored by machine learning methods to discover interesting molecules, before time-consuming experimental synthesizing is attempted. However, these methods often rely on a graph representation, ignoring 3D information necessary for determining the stability of the molecules. We propose a reinforcement learning approach for generating molecules in cartesian coordinates allowing for quantum chemical prediction of the stability. To improve sample-efficiency we learn basic chemical rules from imitation learning on the GDB-11 database to create an initial model applicable for all stoichiometries. We then deploy multiple copies of the model conditioned on a specific stoichiometry in a reinforcement learning setting. The models correctly identify low energy molecules in the database and produce novel isomers not found in the training set. Finally, we apply the model to larger molecules to show how reinforcement learning further refines the imitation learning model in domains far from the training data.
Atomistic Structure Learning Algorithm with surrogate energy model relaxation
Jul 15, 2020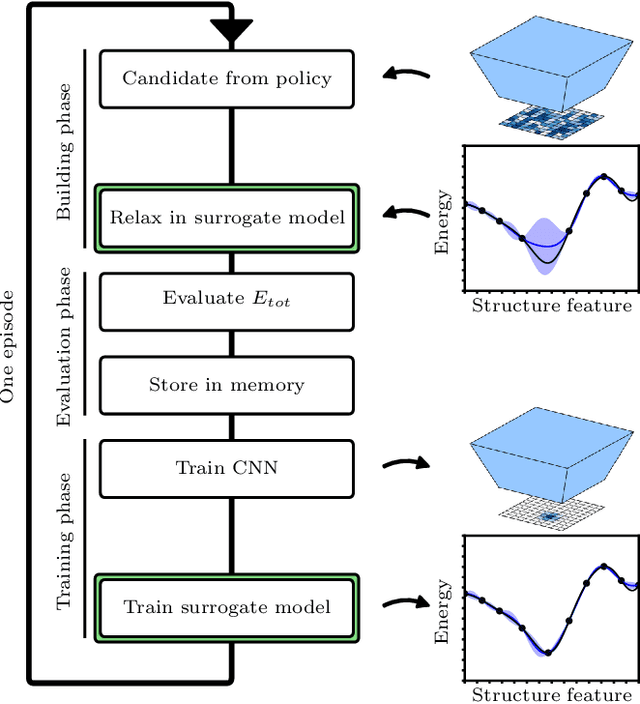
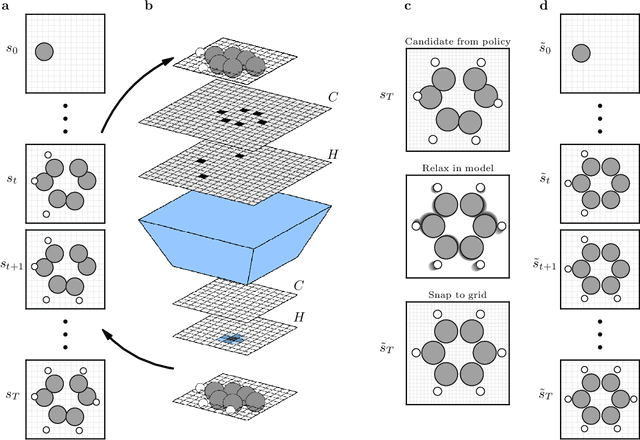
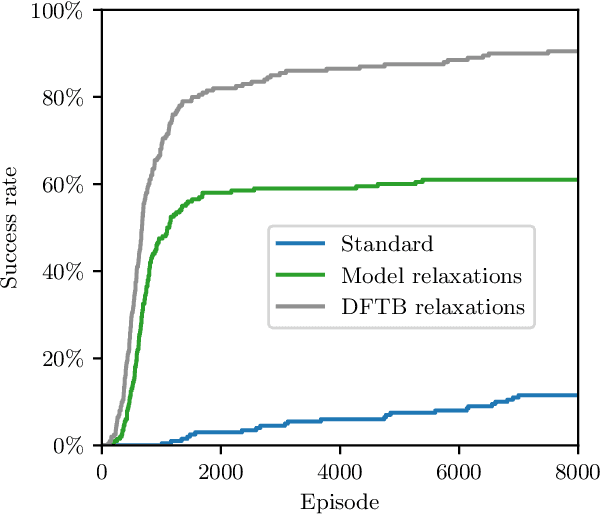
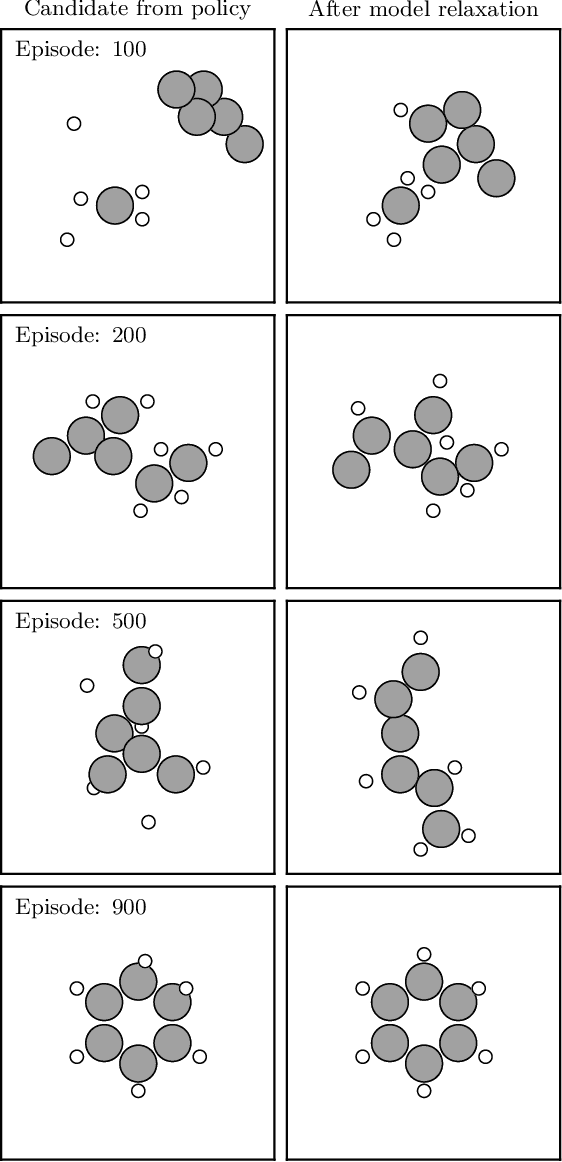
The recently proposed Atomistic Structure Learning Algorithm (ASLA) builds on neural network enabled image recognition and reinforcement learning. It enables fully autonomous structure determination when used in combination with a first-principles total energy calculator, e.g. a density functional theory (DFT) program. To save on the computational requirements, ASLA utilizes the DFT program in a single-point mode, i.e. without allowing for relaxation of the structural candidates according to the force information at the DFT level. In this work, we augment ASLA to establish a surrogate energy model concurrently with its structure search. This enables approximative but computationally cheap relaxation of the structural candidates before the single-point energy evaluation with the computationally expensive DFT program. We demonstrate a significantly increased performance of ASLA for building benzene while utilizing a surrogate energy landscape. Further we apply this model-enhanced ASLA in a thorough investigation of the c(4x8) phase of the Ag(111) surface oxide. ASLA successfully identifies a surface reconstruction which has previously only been guessed on the basis of scanning tunnelling microscopy images.
Atomistic structure learning
Feb 27, 2019
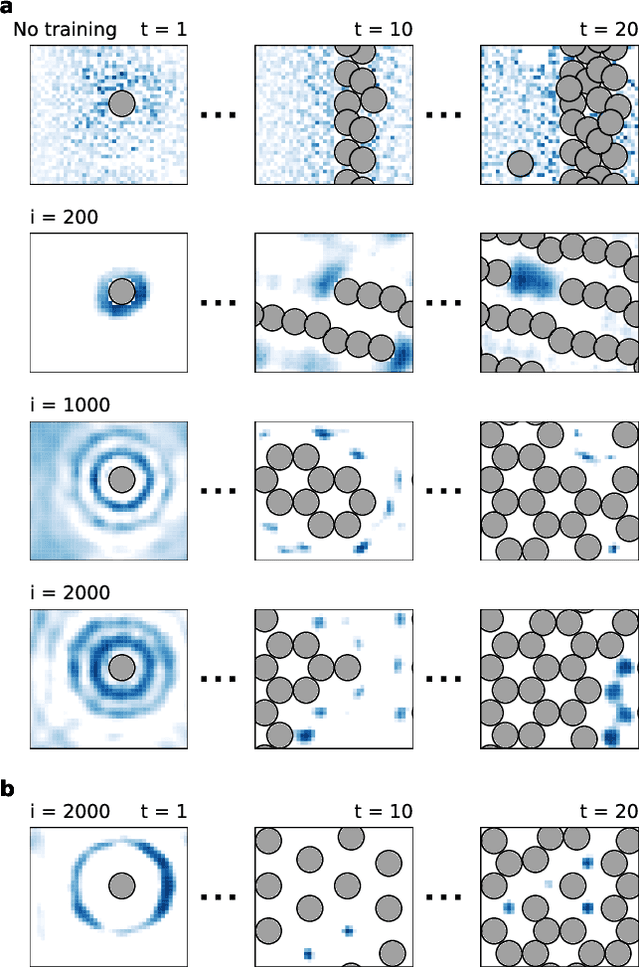
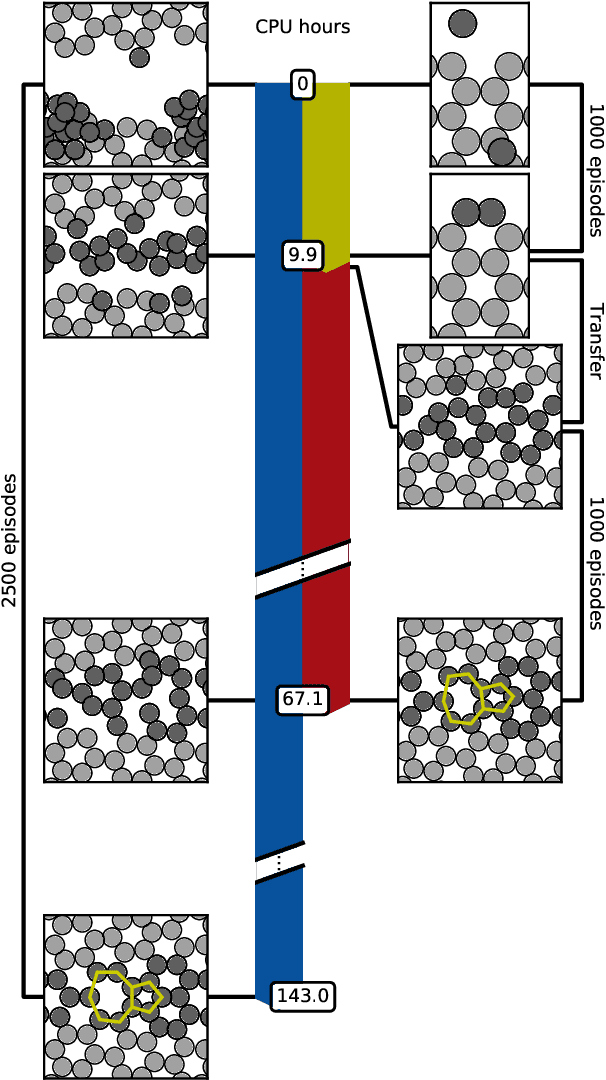

One endeavour of modern physical chemistry is to use bottom-up approaches to design materials and drugs with desired properties. Here we introduce an atomistic structure learning algorithm (ASLA) that utilizes a convolutional neural network to build 2D compounds and layered structures atom by atom. The algorithm takes no prior data or knowledge on atomic interactions but inquires a first-principles quantum mechanical program for physical properties. Using reinforcement learning, the algorithm accumulates knowledge of chemical compound space for a given number and type of atoms and stores this in the neural network, ultimately learning the blueprint for the optimal structural arrangement of the atoms for a given target property. ASLA is demonstrated to work on diverse problems, including grain boundaries in graphene sheets, organic compound formation and a surface oxide structure. This approach to structure prediction is a first step toward direct manipulation of atoms with artificially intelligent first principles computer codes.