 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEvaluating Zero-Shot Scoring for In Vitro Antibody Binding Prediction with Experimental Validation
Dec 07, 2023



The success of therapeutic antibodies relies on their ability to selectively bind antigens. AI-based antibody design protocols have shown promise in generating epitope-specific designs. Many of these protocols use an inverse folding step to generate diverse sequences given a backbone structure. Due to prohibitive screening costs, it is key to identify candidate sequences likely to bind in vitro. Here, we compare the efficacy of 8 common scoring paradigms based on open-source models to classify antibody designs as binders or non-binders. We evaluate these approaches on a novel surface plasmon resonance (SPR) dataset, spanning 5 antigens. Our results show that existing methods struggle to detect binders, and performance is highly variable across antigens. We find that metrics computed on flexibly docked antibody-antigen complexes are more robust, and ensembles scores are more consistent than individual metrics. We provide experimental insight to analyze current scoring techniques, highlighting that the development of robust, zero-shot filters is an important research gap.
Is Transfer Learning Necessary for Protein Landscape Prediction?
Oct 31, 2020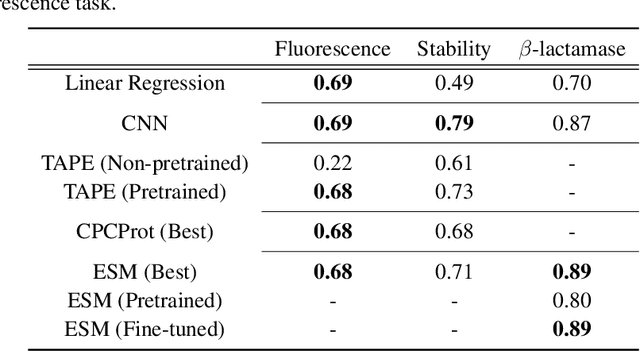
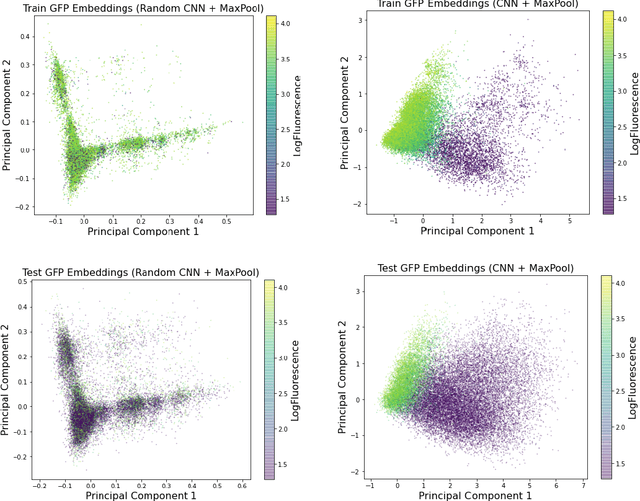
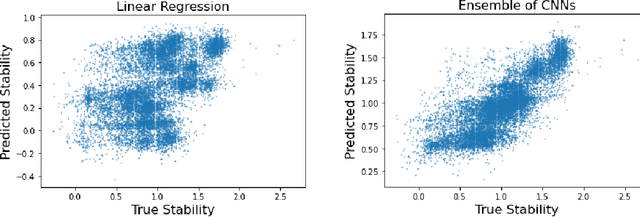
Recently, there has been great interest in learning how to best represent proteins, specifically with fixed-length embeddings. Deep learning has become a popular tool for protein representation learning as a model's hidden layers produce potentially useful vector embeddings. TAPE introduced a number of benchmark tasks and showed that semi-supervised learning, via pretraining language models on a large protein corpus, improved performance on downstream tasks. Two of the tasks (fluorescence prediction and stability prediction) involve learning fitness landscapes. In this paper, we show that CNN models trained solely using supervised learning both compete with and sometimes outperform the best models from TAPE that leverage expensive pretraining on large protein datasets. These CNN models are sufficiently simple and small that they can be trained using a Google Colab notebook. We also find for the fluorescence task that linear regression outperforms our models and the TAPE models. The benchmarking tasks proposed by TAPE are excellent measures of a model's ability to predict protein function and should be used going forward. However, we believe it is important to add baselines from simple models to put the performance of the semi-supervised models that have been reported so far into perspective.
Fixed-Length Protein Embeddings using Contextual Lenses
Oct 15, 2020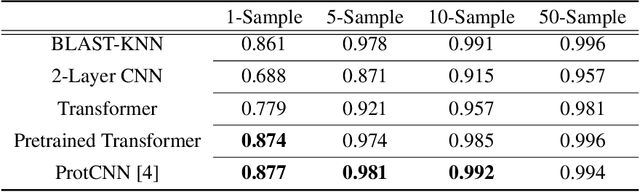
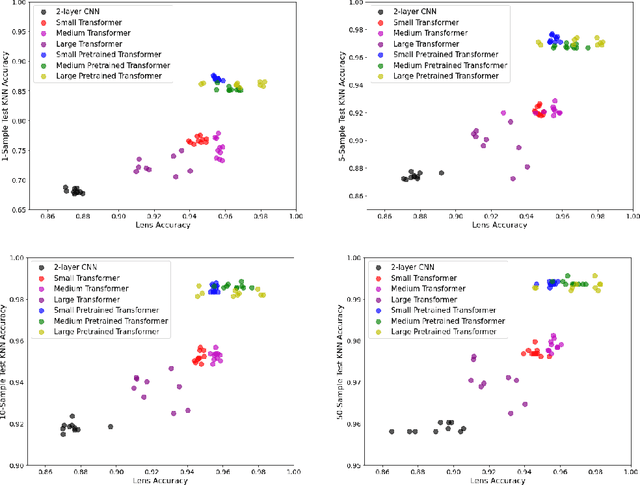
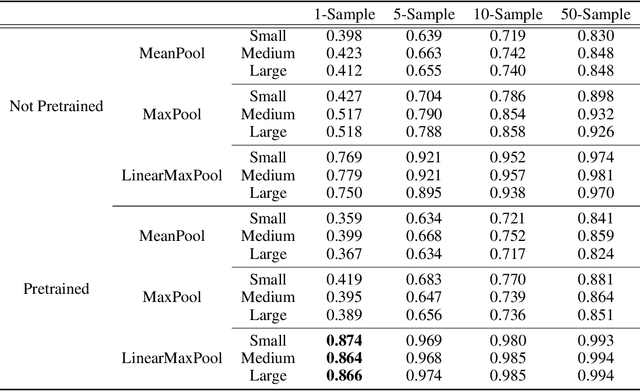
The Basic Local Alignment Search Tool (BLAST) is currently the most popular method for searching databases of biological sequences. BLAST compares sequences via similarity defined by a weighted edit distance, which results in it being computationally expensive. As opposed to working with edit distance, a vector similarity approach can be accelerated substantially using modern hardware or hashing techniques. Such an approach would require fixed-length embeddings for biological sequences. There has been recent interest in learning fixed-length protein embeddings using deep learning models under the hypothesis that the hidden layers of supervised or semi-supervised models could produce potentially useful vector embeddings. We consider transformer (BERT) protein language models that are pretrained on the TrEMBL data set and learn fixed-length embeddings on top of them with contextual lenses. The embeddings are trained to predict the family a protein belongs to for sequences in the Pfam database. We show that for nearest-neighbor family classification, pretraining offers a noticeable boost in performance and that the corresponding learned embeddings are competitive with BLAST. Furthermore, we show that the raw transformer embeddings, obtained via static pooling, do not perform well on nearest-neighbor family classification, which suggests that learning embeddings in a supervised manner via contextual lenses may be a compute-efficient alternative to fine-tuning.