 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGENER: A Parallel Layer Deep Learning Network To Detect Gene-Gene Interactions From Gene Expression Data
Oct 06, 2023Detecting and discovering new gene interactions based on known gene expressions and gene interaction data presents a significant challenge. Various statistical and deep learning methods have attempted to tackle this challenge by leveraging the topological structure of gene interactions and gene expression patterns to predict novel gene interactions. In contrast, some approaches have focused exclusively on utilizing gene expression profiles. In this context, we introduce GENER, a parallel-layer deep learning network designed exclusively for the identification of gene-gene relationships using gene expression data. We conducted two training experiments and compared the performance of our network with that of existing statistical and deep learning approaches. Notably, our model achieved an average AUROC score of 0.834 on the combined BioGRID&DREAM5 dataset, outperforming competing methods in predicting gene-gene interactions.
High-dimensional multi-trait GWAS by reverse prediction of genotypes
Oct 29, 2021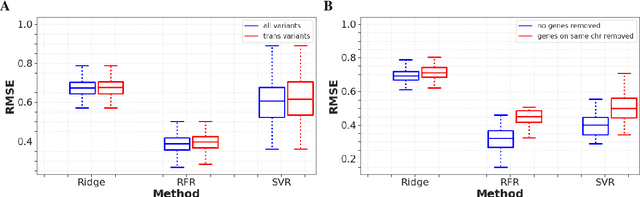
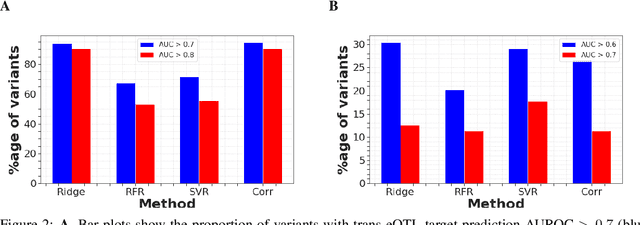
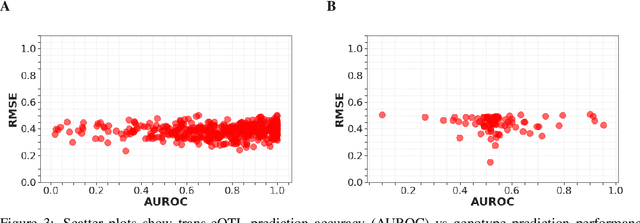
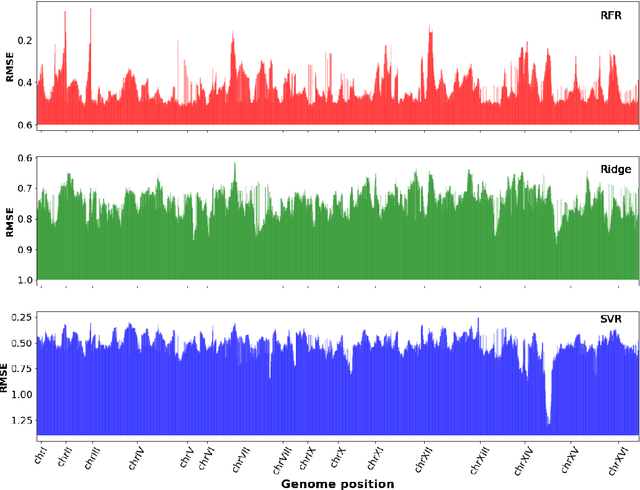
Multi-trait genome-wide association studies (GWAS) use multi-variate statistical methods to identify associations between genetic variants and multiple correlated traits simultaneously, and have higher statistical power than independent univariate analysis of traits. Reverse regression, where genotypes of genetic variants are regressed on multiple traits simultaneously, has emerged as a promising approach to perform multi-trait GWAS in high-dimensional settings where the number of traits exceeds the number of samples. We extended this approach and analyzed different machine learning methods (ridge regression, random forests and support vector machines)for reverse regression in multi-trait GWAS, using genotypes, gene expression data and ground-truth transcriptional regulatory networks from the DREAM5 SysGen Challenge and from a cross between two yeast strains to evaluate methods. We found that genotype prediction performance, in terms of root mean squared error (RMSE), allowed to distinguish between genomic regions with high and low transcriptional activity. Moreover, model feature coefficients correlated with the strength of association between variants and individual traits, and were predictive of true trans-eQTL target genes, with complementary findings across methods.