 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWhat Ails Generative Structure-based Drug Design: Too Little or Too Much Expressivity?
Paper and Code

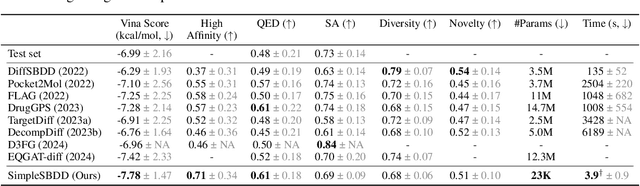

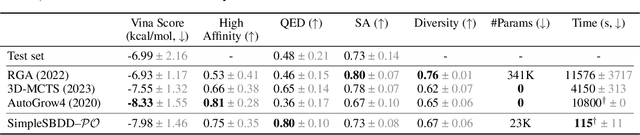
Several generative models with elaborate training and sampling procedures have been proposed recently to accelerate structure-based drug design (SBDD); however, perplexingly, their empirical performance turns out to be suboptimal. We seek to better understand this phenomenon from both theoretical and empirical perspectives. Since most of these models apply graph neural networks (GNNs), one may suspect that they inherit the representational limitations of GNNs. We analyze this aspect, establishing the first such results for protein-ligand complexes. A plausible counterview may attribute the underperformance of these models to their excessive parameterizations, inducing expressivity at the expense of generalization. We also investigate this possibility with a simple metric-aware approach that learns an economical surrogate for affinity to infer an unlabelled molecular graph and optimizes for labels conditioned on this graph and molecular properties. The resulting model achieves state-of-the-art results using 100x fewer trainable parameters and affords up to 1000x speedup. Collectively, our findings underscore the need to reassess and redirect the existing paradigm and efforts for SBDD.