 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVAMPnets: Deep learning of molecular kinetics
Paper and Code

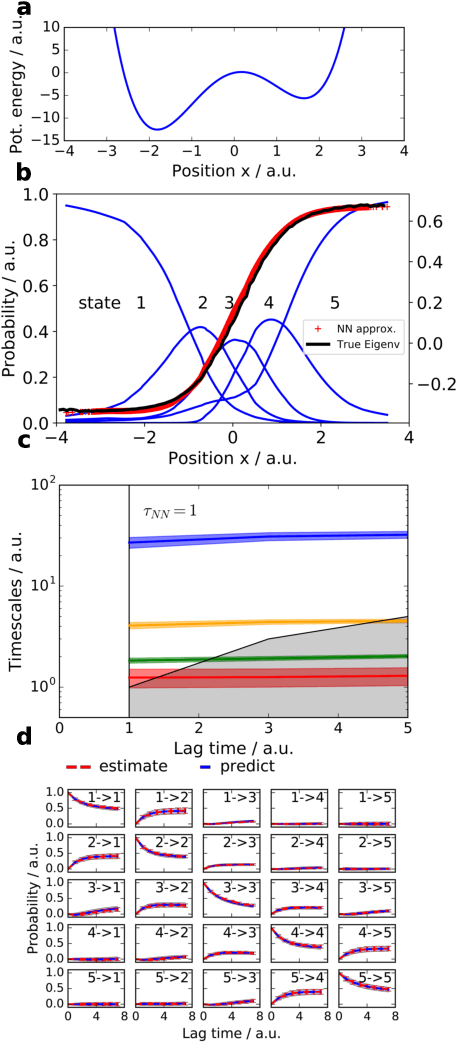


There is an increasing demand for computing the relevant structures, equilibria and long-timescale kinetics of biomolecular processes, such as protein-drug binding, from high-throughput molecular dynamics simulations. Current methods employ transformation of simulated coordinates into structural features, dimension reduction, clustering the dimension-reduced data, and estimation of a Markov state model or related model of the interconversion rates between molecular structures. This handcrafted approach demands a substantial amount of modeling expertise, as poor decisions at any step will lead to large modeling errors. Here we employ the variational approach for Markov processes (VAMP) to develop a deep learning framework for molecular kinetics using neural networks, dubbed VAMPnets. A VAMPnet encodes the entire mapping from molecular coordinates to Markov states, thus combining the whole data processing pipeline in a single end-to-end framework. Our method performs equally or better than state-of-the art Markov modeling methods and provides easily interpretable few-state kinetic models.