 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUtilizing Mutations to Evaluate Interpretability of Neural Networks on Genomic Data
Paper and Code
Dec 12, 2022
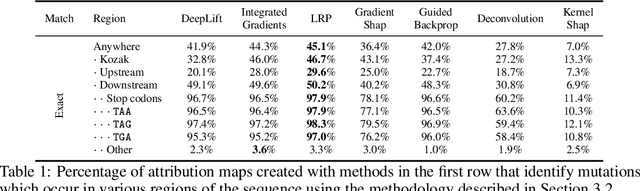
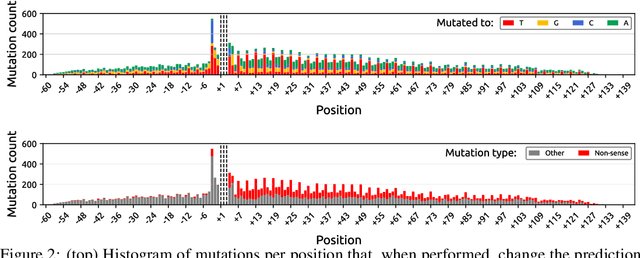

Even though deep neural networks (DNNs) achieve state-of-the-art results for a number of problems involving genomic data, getting DNNs to explain their decision-making process has been a major challenge due to their black-box nature. One way to get DNNs to explain their reasoning for prediction is via attribution methods which are assumed to highlight the parts of the input that contribute to the prediction the most. Given the existence of numerous attribution methods and a lack of quantitative results on the fidelity of those methods, selection of an attribution method for sequence-based tasks has been mostly done qualitatively. In this work, we take a step towards identifying the most faithful attribution method by proposing a computational approach that utilizes point mutations. Providing quantitative results on seven popular attribution methods, we find Layerwise Relevance Propagation (LRP) to be the most appropriate one for translation initiation, with LRP identifying two important biological features for translation: the integrity of Kozak sequence as well as the detrimental effects of premature stop codons.