 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUsing Graph Neural Networks for Mass Spectrometry Prediction
Paper and Code
Oct 09, 2020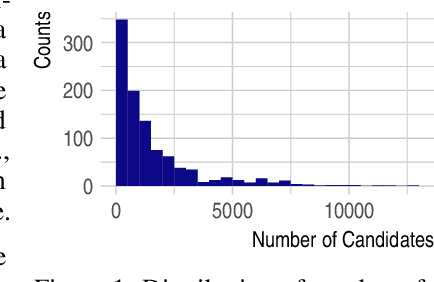
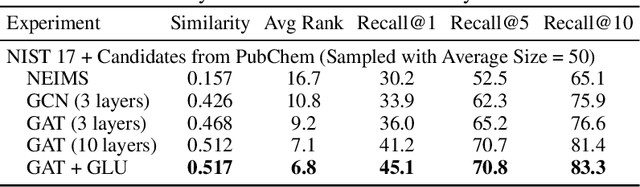
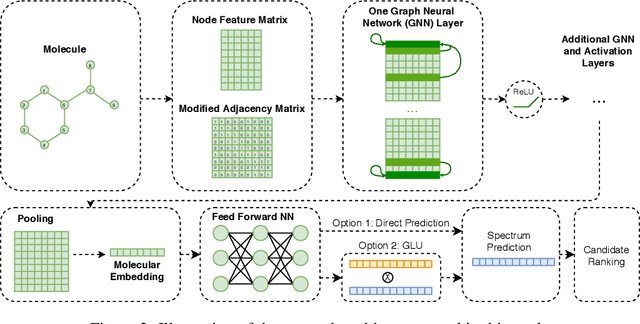
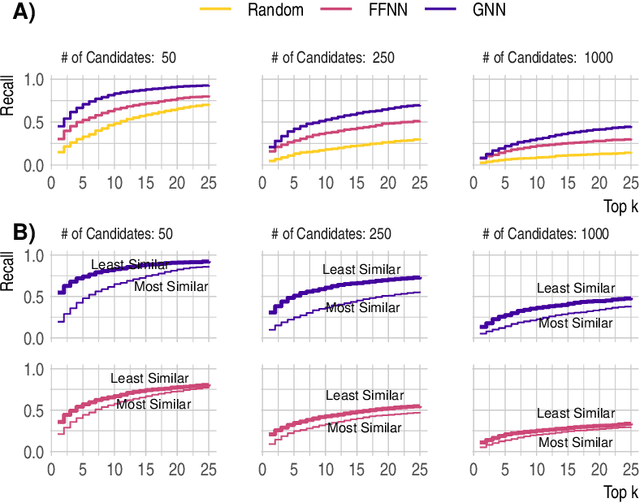
Detecting and quantifying products of cellular metabolism using Mass Spectrometry (MS) has already shown great promise in many biological and biomedical applications. The biggest challenge in metabolomics is annotation, where measured spectra are assigned chemical identities. Despite advances, current methods provide limited annotation for measured spectra. Here, we explore using graph neural networks (GNNs) to predict the spectra. The input to our model is a molecular graph. The model is trained and tested on the NIST 17 LC-MS dataset. We compare our results to NEIMS, a neural network model that utilizes molecular fingerprints as inputs. Our results show that GNN-based models offer higher performance than NEIMS. Importantly, we show that ranking results heavily depend on the candidate set size and on the similarity of the candidates to the target molecule, thus highlighting the need for consistent, well-characterized evaluation protocols for this domain.