 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTopological Descriptors Help Predict Guest Adsorption in Nanoporous Materials
Paper and Code


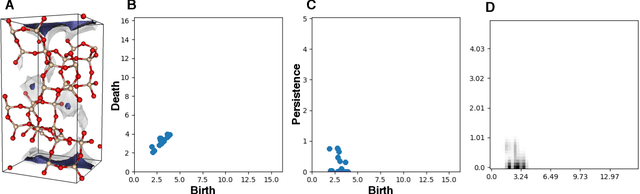
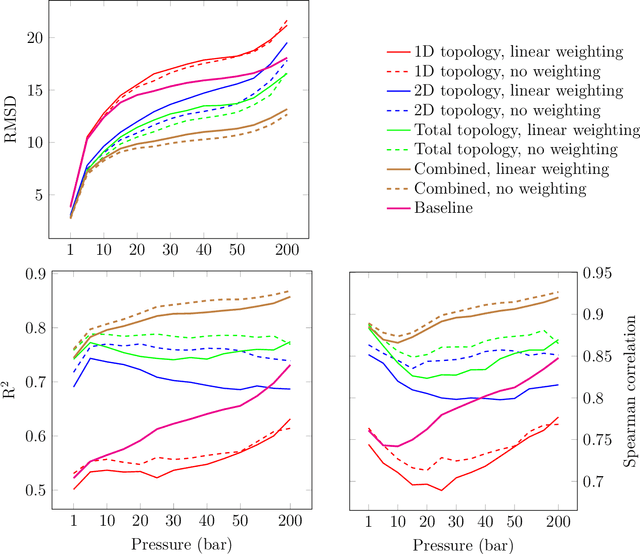
Machine learning has emerged as an attractive alternative to experiments and simulations for predicting material properties. Usually, such an approach relies on specific domain knowledge for feature design: each learning target requires careful selection of features that an expert recognizes as important for the specific task. The major drawback of this approach is that computation of only a few structural features has been implemented so far, and it is difficult to tell a priori which features are important for a particular application. The latter problem has been empirically observed for predictors of guest uptake in nanoporous materials: local and global porosity features become dominant descriptors at low and high pressures, respectively. We investigate a feature representation of materials using tools from topological data analysis. Specifically, we use persistent homology to describe the geometry of nanoporous materials at various scales. We combine our topological descriptor with traditional structural features and investigate the relative importance of each to the prediction tasks. We demonstrate an application of this feature representation by predicting methane adsorption in zeolites, for pressures in the range of 1-200 bar. Our results not only show a considerable improvement compared to the baseline, but they also highlight that topological features capture information complementary to the structural features, which is especially important for the adsorption at low pressure, a task particularly difficult for the traditional features.