 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQuantitative Evaluation of Explainable Graph Neural Networks for Molecular Property Prediction
Paper and Code
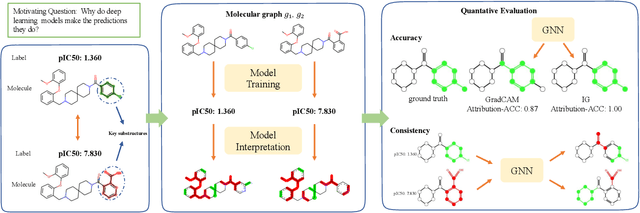
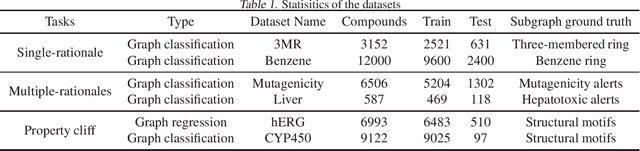
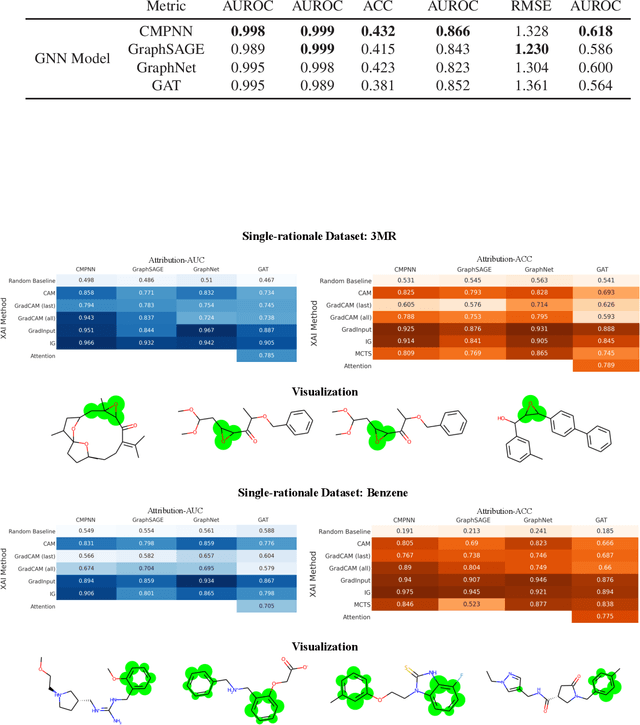
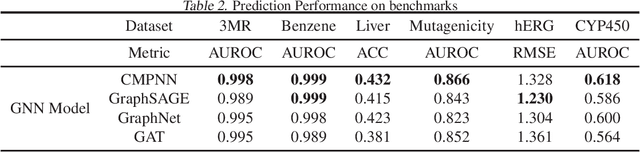
Advances in machine learning have led to graph neural network-based methods for drug discovery, yielding promising results in molecular design, chemical synthesis planning, and molecular property prediction. However, current graph neural networks (GNNs) remain of limited acceptance in drug discovery is limited due to their lack of interpretability. Although this major weakness has been mitigated by the development of explainable artificial intelligence (XAI) techniques, the "ground truth" assignment in most explainable tasks ultimately rests with subjective judgments by humans so that the quality of model interpretation is hard to evaluate in quantity. In this work, we first build three levels of benchmark datasets to quantitatively assess the interpretability of the state-of-the-art GNN models. Then we implemented recent XAI methods in combination with different GNN algorithms to highlight the benefits, limitations, and future opportunities for drug discovery. As a result, GradInput and IG generally provide the best model interpretability for GNNs, especially when combined with GraphNet and CMPNN. The integrated and developed XAI package is fully open-sourced and can be used by practitioners to train new models on other drug discovery tasks.