 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQuantifying homologous proteins and proteoforms
Paper and Code
Aug 05, 2017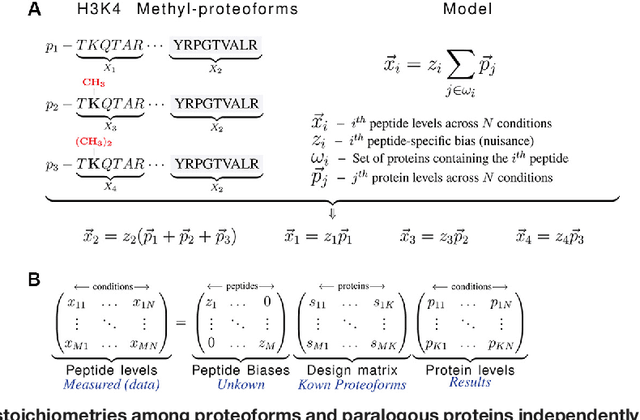
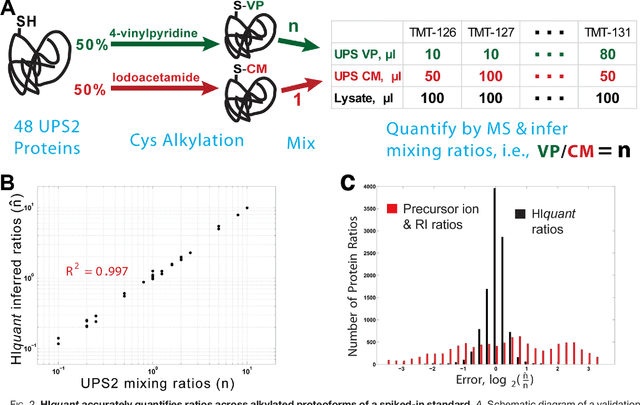
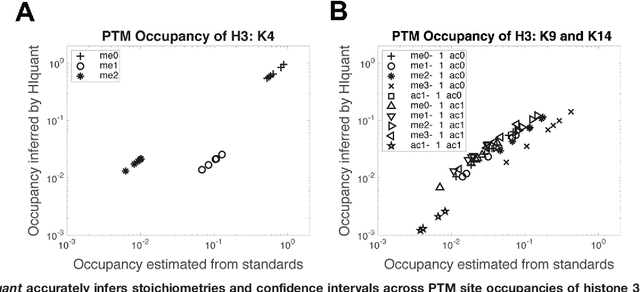
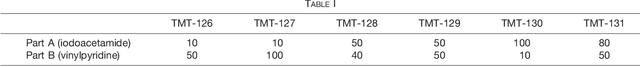
Many proteoforms - arising from alternative splicing, post-translational modifications (PTMs), or paralogous genes - have distinct biological functions, such as histone PTM proteoforms. However, their quantification by existing bottom-up mass-spectrometry (MS) methods is undermined by peptide-specific biases. To avoid these biases, we developed and implemented a first-principles model (HIquant) for quantifying proteoform stoichiometries. We characterized when MS data allow inferring proteoform stoichiometries by HIquant, derived an algorithm for optimal inference, and demonstrated experimentally high accuracy in quantifying fractional PTM occupancy without using external standards, even in the challenging case of the histone modification code. HIquant server is implemented at: https://web.northeastern.edu/slavov/2014_HIquant/