 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMultiModal-Learning for Predicting Molecular Properties: A Framework Based on Image and Graph Structures
Paper and Code
Nov 28, 2023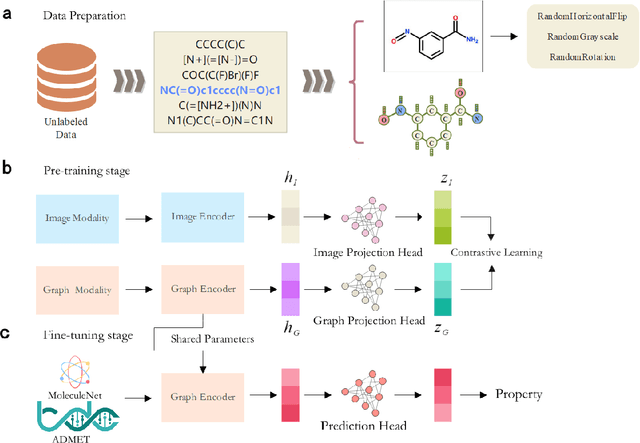
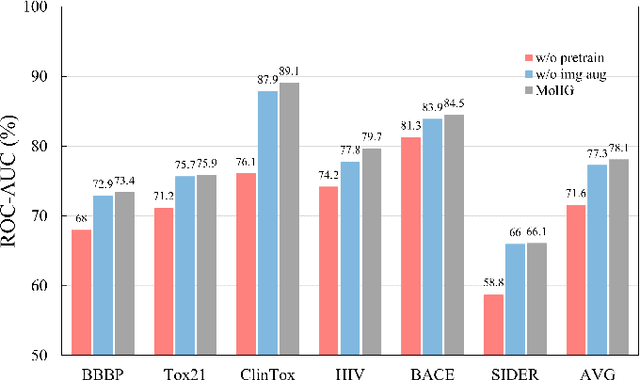
The quest for accurate prediction of drug molecule properties poses a fundamental challenge in the realm of Artificial Intelligence Drug Discovery (AIDD). An effective representation of drug molecules emerges as a pivotal component in this pursuit. Contemporary leading-edge research predominantly resorts to self-supervised learning (SSL) techniques to extract meaningful structural representations from large-scale, unlabeled molecular data, subsequently fine-tuning these representations for an array of downstream tasks. However, an inherent shortcoming of these studies lies in their singular reliance on one modality of molecular information, such as molecule image or SMILES representations, thus neglecting the potential complementarity of various molecular modalities. In response to this limitation, we propose MolIG, a novel MultiModaL molecular pre-training framework for predicting molecular properties based on Image and Graph structures. MolIG model innovatively leverages the coherence and correlation between molecule graph and molecule image to execute self-supervised tasks, effectively amalgamating the strengths of both molecular representation forms. This holistic approach allows for the capture of pivotal molecular structural characteristics and high-level semantic information. Upon completion of pre-training, Graph Neural Network (GNN) Encoder is used for the prediction of downstream tasks. In comparison to advanced baseline models, MolIG exhibits enhanced performance in downstream tasks pertaining to molecular property prediction within benchmark groups such as MoleculeNet Benchmark Group and ADMET Benchmark Group.