 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular Design Based on Integer Programming and Splitting Data Sets by Hyperplanes
Paper and Code
Apr 27, 2023
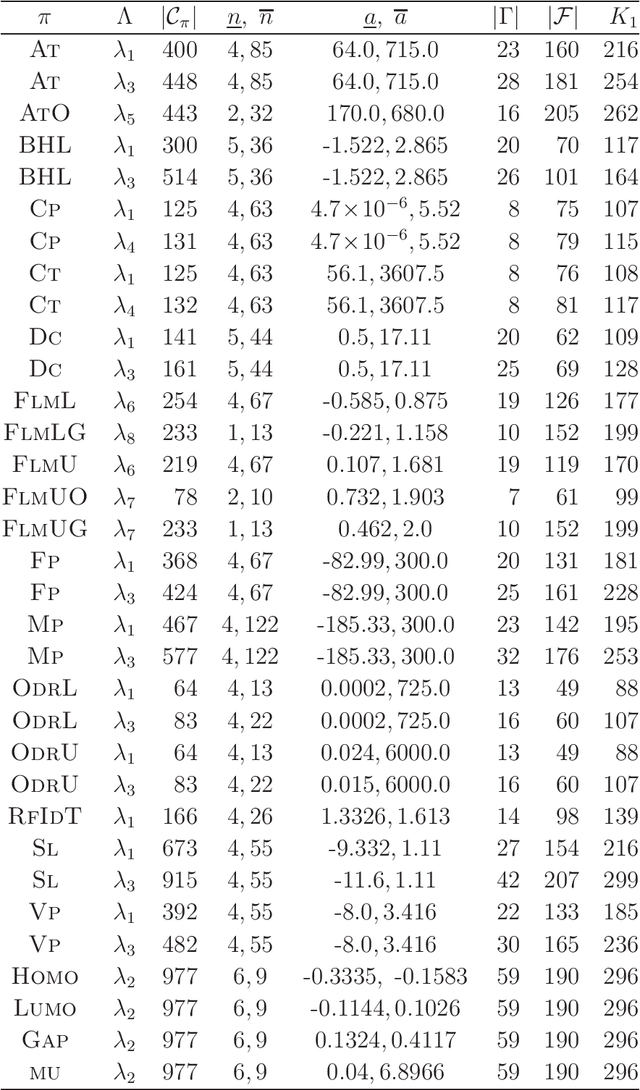

A novel framework for designing the molecular structure of chemical compounds with a desired chemical property has recently been proposed. The framework infers a desired chemical graph by solving a mixed integer linear program (MILP) that simulates the computation process of a feature function defined by a two-layered model on chemical graphs and a prediction function constructed by a machine learning method. To improve the learning performance of prediction functions in the framework, we design a method that splits a given data set $\mathcal{C}$ into two subsets $\mathcal{C}^{(i)},i=1,2$ by a hyperplane in a chemical space so that most compounds in the first (resp., second) subset have observed values lower (resp., higher) than a threshold $\theta$. We construct a prediction function $\psi$ to the data set $\mathcal{C}$ by combining prediction functions $\psi_i,i=1,2$ each of which is constructed on $\mathcal{C}^{(i)}$ independently. The results of our computational experiments suggest that the proposed method improved the learning performance for several chemical properties to which a good prediction function has been difficult to construct.