 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMachine Learning Implicit Solvation for Molecular Dynamics
Paper and Code
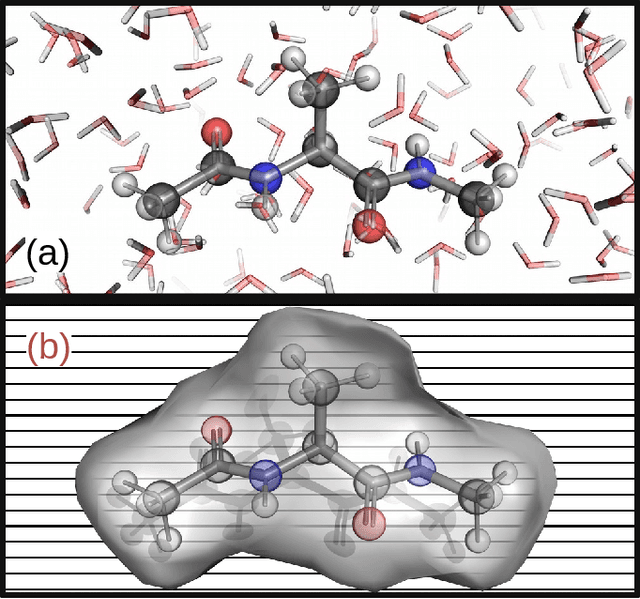
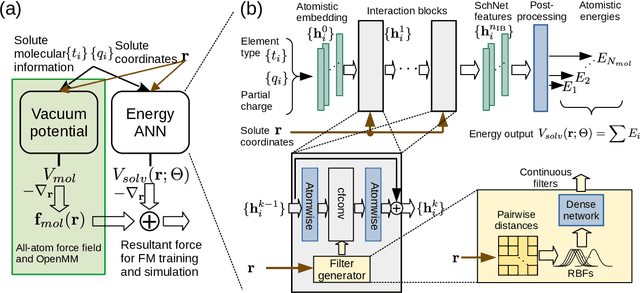
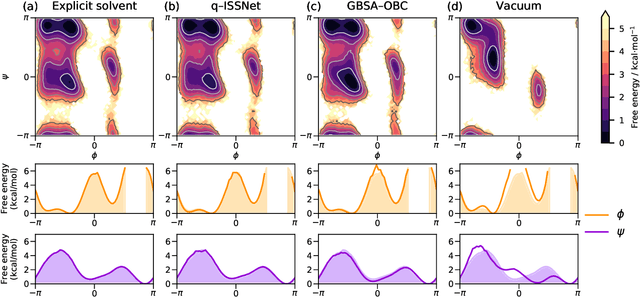
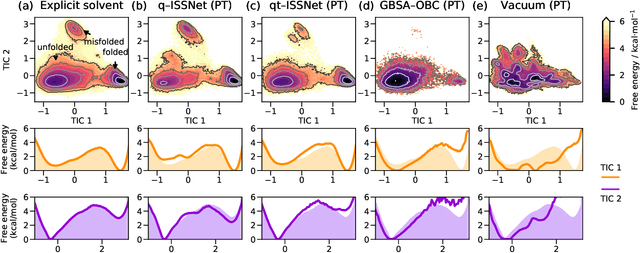
Accurate modeling of the solvent environment for biological molecules is crucial for computational biology and drug design. A popular approach to achieve long simulation time scales for large system sizes is to incorporate the effect of the solvent in a mean-field fashion with implicit solvent models. However, a challenge with existing implicit solvent models is that they often lack accuracy or certain physical properties compared to explicit solvent models, as the many-body effects of the neglected solvent molecules is difficult to model as a mean field. Here, we leverage machine learning (ML) and multi-scale coarse graining (CG) in order to learn implicit solvent models that can approximate the energetic and thermodynamic properties of a given explicit solvent model with arbitrary accuracy, given enough training data. Following the previous ML--CG models CGnet and CGSchnet, we introduce ISSNet, a graph neural network, to model the implicit solvent potential of mean force. ISSNet can learn from explicit solvent simulation data and be readily applied to MD simulations. We compare the solute conformational distributions under different solvation treatments for two peptide systems. The results indicate that ISSNet models can outperform widely-used generalized Born and surface area models in reproducing the thermodynamics of small protein systems with respect to explicit solvent. The success of this novel method demonstrates the potential benefit of applying machine learning methods in accurate modeling of solvent effects for in silico research and biomedical applications.