 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeKPGT: Knowledge-Guided Pre-training of Graph Transformer for Molecular Property Prediction
Paper and Code
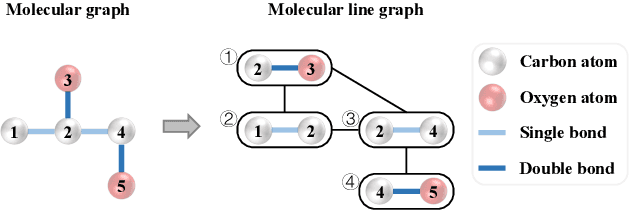
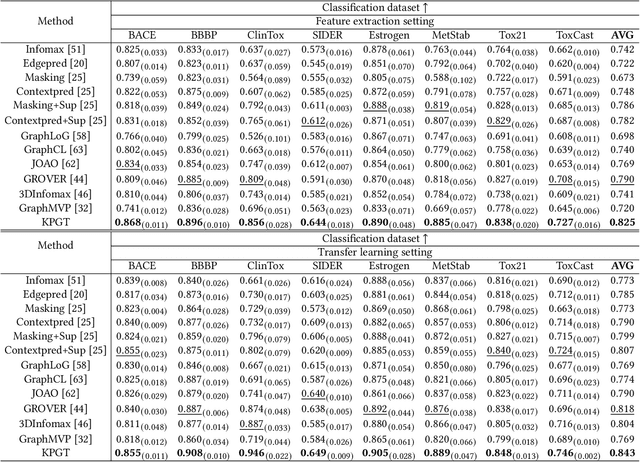


Designing accurate deep learning models for molecular property prediction plays an increasingly essential role in drug and material discovery. Recently, due to the scarcity of labeled molecules, self-supervised learning methods for learning generalizable and transferable representations of molecular graphs have attracted lots of attention. In this paper, we argue that there exist two major issues hindering current self-supervised learning methods from obtaining desired performance on molecular property prediction, that is, the ill-defined pre-training tasks and the limited model capacity. To this end, we introduce Knowledge-guided Pre-training of Graph Transformer (KPGT), a novel self-supervised learning framework for molecular graph representation learning, to alleviate the aforementioned issues and improve the performance on the downstream molecular property prediction tasks. More specifically, we first introduce a high-capacity model, named Line Graph Transformer (LiGhT), which emphasizes the importance of chemical bonds and is mainly designed to model the structural information of molecular graphs. Then, a knowledge-guided pre-training strategy is proposed to exploit the additional knowledge of molecules to guide the model to capture the abundant structural and semantic information from large-scale unlabeled molecular graphs. Extensive computational tests demonstrated that KPGT can offer superior performance over current state-of-the-art methods on several molecular property prediction tasks.