 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproving Compound Activity Classification via Deep Transfer and Representation Learning
Paper and Code



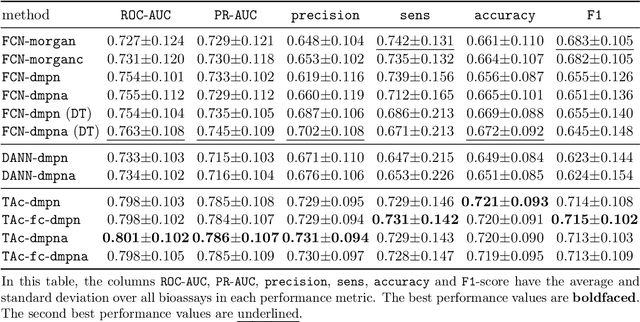
Recent advances in molecular machine learning, especially deep neural networks such as Graph Neural Networks (GNNs) for predicting structure activity relationships (SAR) have shown tremendous potential in computer-aided drug discovery. However, the applicability of such deep neural networks are limited by the requirement of large amounts of training data. In order to cope with limited training data for a target task, transfer learning for SAR modeling has been recently adopted to leverage information from data of related tasks. In this work, in contrast to the popular parameter-based transfer learning such as pretraining, we develop novel deep transfer learning methods TAc and TAc-fc to leverage source domain data and transfer useful information to the target domain. TAc learns to generate effective molecular features that can generalize well from one domain to another, and increase the classification performance in the target domain. Additionally, TAc-fc extends TAc by incorporating novel components to selectively learn feature-wise and compound-wise transferability. We used the bioassay screening data from PubChem, and identified 120 pairs of bioassays such that the active compounds in each pair are more similar to each other compared to its inactive compounds. Overall, TAc achieves the best performance with average ROC-AUC of 0.801; it significantly improves ROC-AUC of 83% target tasks with average task-wise performance improvement of 7.102%, compared to the best baseline FCN-dmpna (DT). Our experiments clearly demonstrate that TAc achieves significant improvement over all baselines across a large number of target tasks. Furthermore, although TAc-fc achieves slightly worse ROC-AUC on average compared to TAc (0.798 vs 0.801), TAc-fc still achieves the best performance on more tasks in terms of PR-AUC and F1 compared to other methods.