 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnergy-based models for atomic-resolution protein conformations
Paper and Code
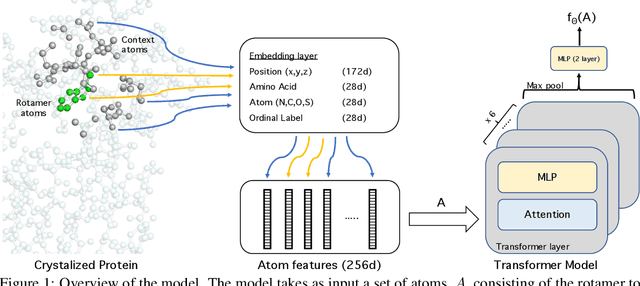
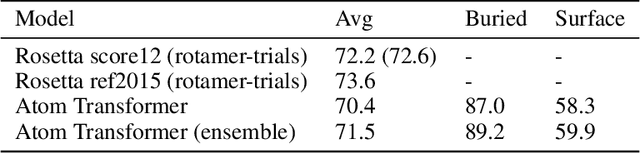
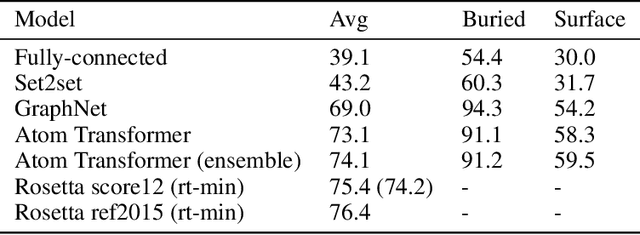
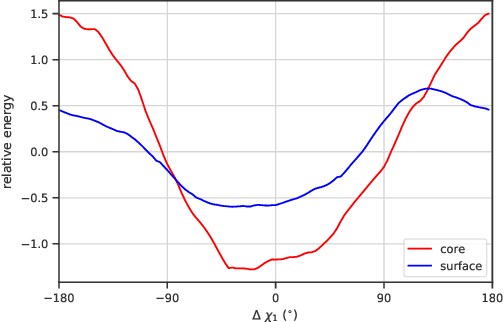
We propose an energy-based model (EBM) of protein conformations that operates at atomic scale. The model is trained solely on crystallized protein data. By contrast, existing approaches for scoring conformations use energy functions that incorporate knowledge of physical principles and features that are the complex product of several decades of research and tuning. To evaluate the model, we benchmark on the rotamer recovery task, the problem of predicting the conformation of a side chain from its context within a protein structure, which has been used to evaluate energy functions for protein design. The model achieves performance close to that of the Rosetta energy function, a state-of-the-art method widely used in protein structure prediction and design. An investigation of the model's outputs and hidden representations finds that it captures physicochemical properties relevant to protein energy.