 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEmbed-Search-Align: DNA Sequence Alignment using Transformer Models
Paper and Code
Sep 20, 2023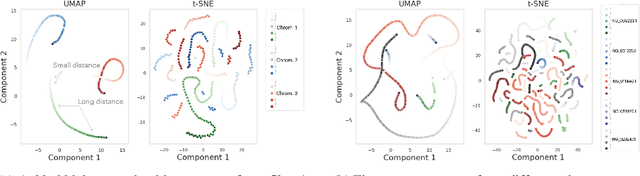
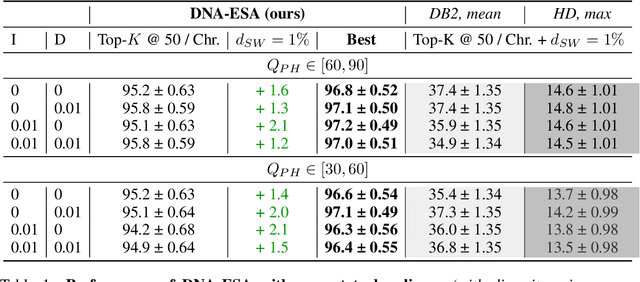
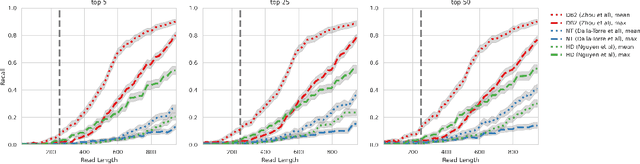
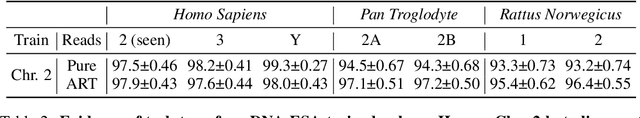
DNA sequence alignment involves assigning short DNA reads to the most probable locations on an extensive reference genome. This process is crucial for various genomic analyses, including variant calling, transcriptomics, and epigenomics. Conventional methods, refined over decades, tackle this challenge in two steps: genome indexing followed by efficient search to locate likely positions for given reads. Building on the success of Large Language Models (LLM) in encoding text into embeddings, where the distance metric captures semantic similarity, recent efforts have explored whether the same Transformer architecture can produce numerical representations for DNA sequences. Such models have shown early promise in tasks involving classification of short DNA sequences, such as the detection of coding vs non-coding regions, as well as the identification of enhancer and promoter sequences. Performance at sequence classification tasks does not, however, translate to sequence alignment, where it is necessary to conduct a genome-wide search to successfully align every read. We address this open problem by framing it as an Embed-Search-Align task. In this framework, a novel encoder model DNA-ESA generates representations of reads and fragments of the reference, which are projected into a shared vector space where the read-fragment distance is used as surrogate for alignment. In particular, DNA-ESA introduces: (1) Contrastive loss for self-supervised training of DNA sequence representations, facilitating rich sequence-level embeddings, and (2) a DNA vector store to enable search across fragments on a global scale. DNA-ESA is >97% accurate when aligning 250-length reads onto a human reference genome of 3 gigabases (single-haploid), far exceeds the performance of 6 recent DNA-Transformer model baselines and shows task transfer across chromosomes and species.