 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficiently predicting high resolution mass spectra with graph neural networks
Paper and Code
Jan 26, 2023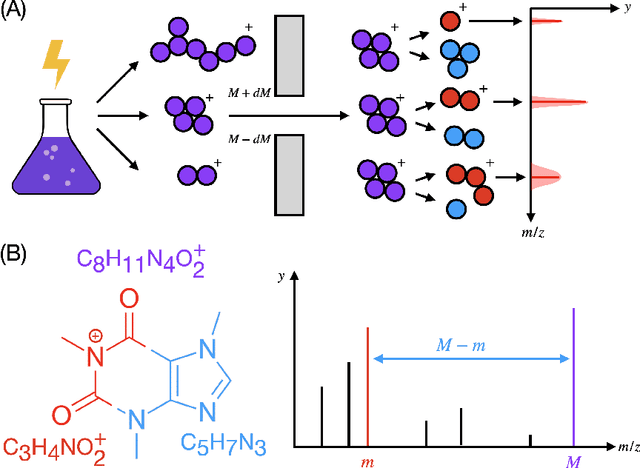

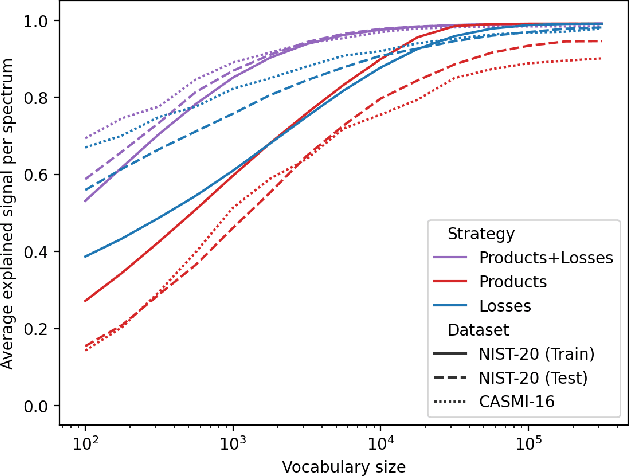

Identifying a small molecule from its mass spectrum is the primary open problem in computational metabolomics. This is typically cast as information retrieval: an unknown spectrum is matched against spectra predicted computationally from a large database of chemical structures. However, current approaches to spectrum prediction model the output space in ways that force a tradeoff between capturing high resolution mass information and tractable learning. We resolve this tradeoff by casting spectrum prediction as a mapping from an input molecular graph to a probability distribution over molecular formulas. We discover that a large corpus of mass spectra can be closely approximated using a fixed vocabulary constituting only 2% of all observed formulas. This enables efficient spectrum prediction using an architecture similar to graph classification - GrAFF-MS - achieving significantly lower prediction error and orders-of-magnitude faster runtime than state-of-the-art methods.