 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDPST: De Novo Peptide Sequencing with Amino-Acid-Aware Transformers
Paper and Code
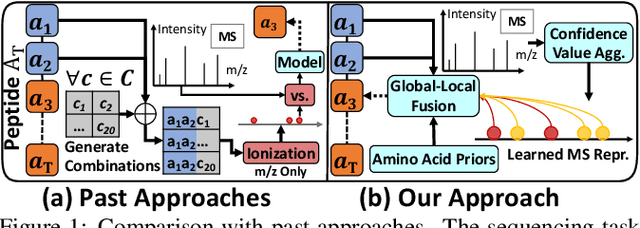



De novo peptide sequencing aims to recover amino acid sequences of a peptide from tandem mass spectrometry (MS) data. Existing approaches for de novo analysis enumerate MS evidence for all amino acid classes during inference. It leads to over-trimming on receptive fields of MS data and restricts MS evidence associated with following undecoded amino acids. Our approach, DPST, circumvents these limitations with two key components: (1) A confidence value aggregation encoder to sketch spectrum representations according to amino-acid-based connectivity among MS; (2) A global-local fusion decoder to progressively assimilate contextualized spectrum representations with a predefined preconception of localized MS evidence and amino acid priors. Our components originate from a closed-form solution and selectively attend to informative amino-acid-aware MS representations. Through extensive empirical studies, we demonstrate the superiority of DPST, showing that it outperforms state-of-the-art approaches by a margin of 12% - 19% peptide accuracy.