Get our free extension to see links to code for papers anywhere online!Free add-on: code for papers everywhere!Free add-on: See code for papers anywhere!
 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeePKS: a comprehensive data-driven approach towards chemically accurate density functional theory
Paper and Code
Aug 01, 2020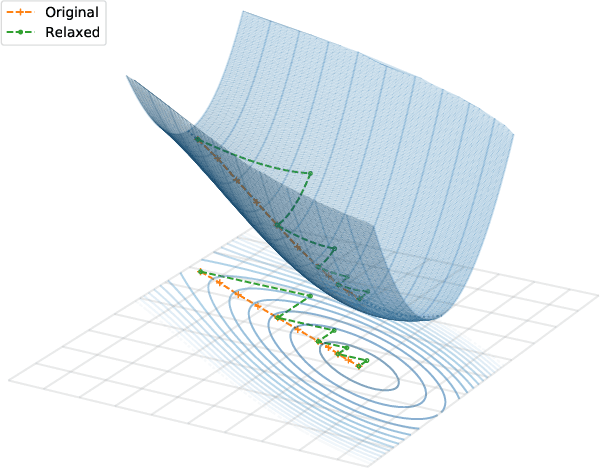
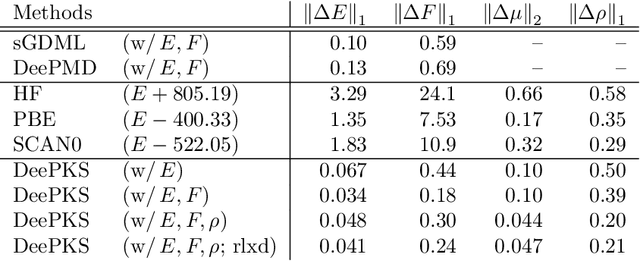
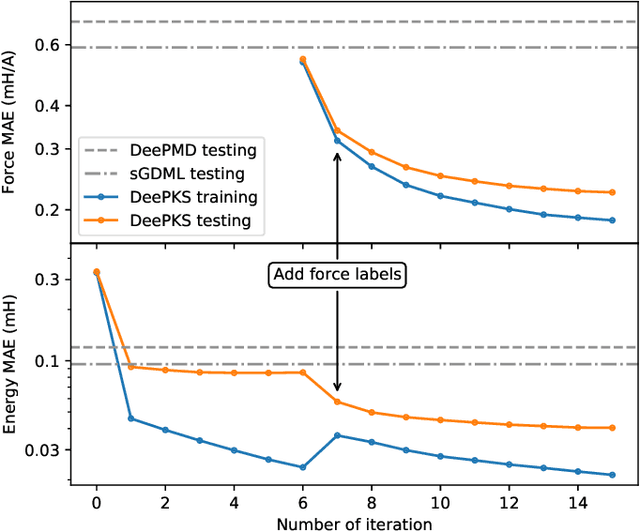
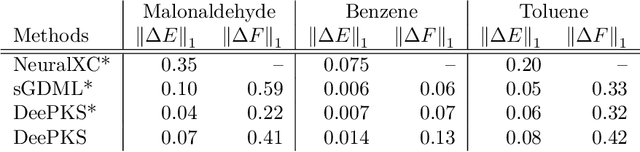
We propose a general machine learning-based framework for building an accurate and widely-applicable energy functional within the framework of generalized Kohn-Sham density functional theory. To this end, we develop a way of training self-consistent models that are capable of taking large datasets from different systems and different kinds of labels. We demonstrate that the functional that results from this training procedure gives chemically accurate predictions on energy, force, dipole, and electron density for a large class of molecules. It can be continuously improved when more and more data are available.
View paper on