Get our free extension to see links to code for papers anywhere online!Free add-on: code for papers everywhere!Free add-on: See code for papers anywhere!
 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeData Requirement for Phylogenetic Inference from Multiple Loci: A New Distance Method
Paper and Code
Jun 30, 2014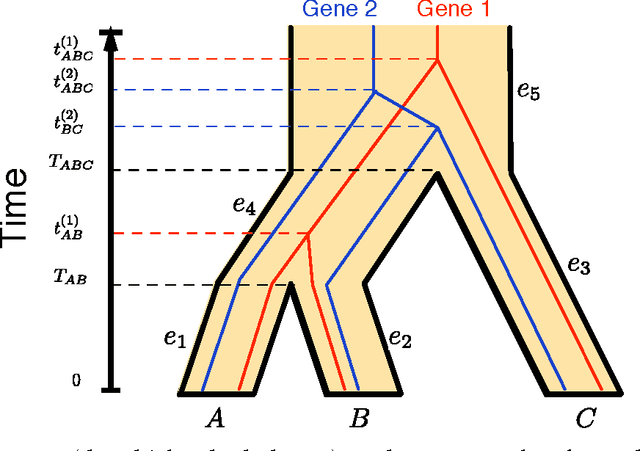

We consider the problem of estimating the evolutionary history of a set of species (phylogeny or species tree) from several genes. It is known that the evolutionary history of individual genes (gene trees) might be topologically distinct from each other and from the underlying species tree, possibly confounding phylogenetic analysis. A further complication in practice is that one has to estimate gene trees from molecular sequences of finite length. We provide the first full data-requirement analysis of a species tree reconstruction method that takes into account estimation errors at the gene level. Under that criterion, we also devise a novel reconstruction algorithm that provably improves over all previous methods in a regime of interest.
* 19 pages, 2 figures. Preliminary version to appear in IEEE ISIT 2014.
Added acknowledgements and made the proof of the "equality" part of Theorem 3
explicit in Appendix C
View paper on