 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAssessing Uncertainties in X-ray Single-particle Three-dimensional reconstructions
Paper and Code
Jan 02, 2017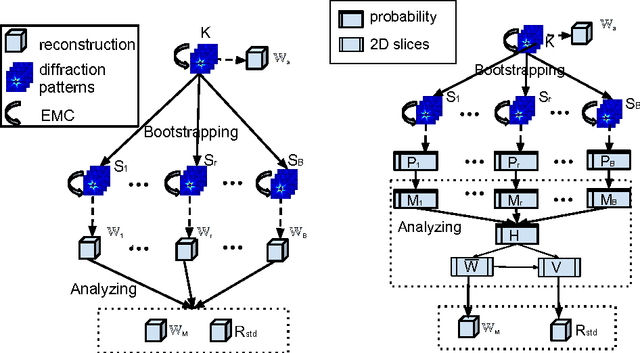
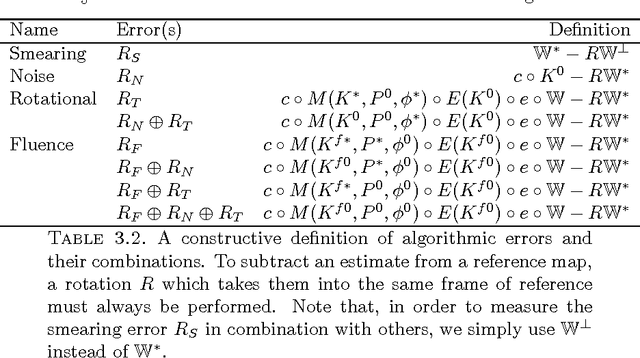
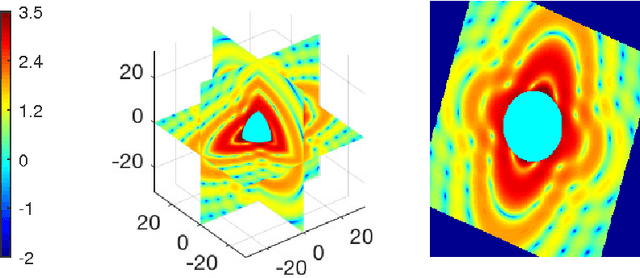
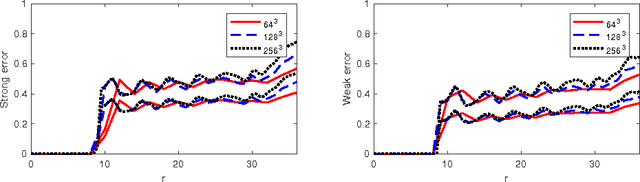
Modern technology for producing extremely bright and coherent X-ray laser pulses provides the possibility to acquire a large number of diffraction patterns from individual biological nanoparticles, including proteins, viruses, and DNA. These two-dimensional diffraction patterns can be practically reconstructed and retrieved down to a resolution of a few \angstrom. In principle, a sufficiently large collection of diffraction patterns will contain the required information for a full three-dimensional reconstruction of the biomolecule. The computational methodology for this reconstruction task is still under development and highly resolved reconstructions have not yet been produced. We analyze the Expansion-Maximization-Compression scheme, the current state of the art approach for this very challenging application, by isolating different sources of uncertainty. Through numerical experiments on synthetic data we evaluate their respective impact. We reach conclusions of relevance for handling actual experimental data, as well as pointing out certain improvements to the underlying estimation algorithm. We also introduce a practically applicable computational methodology in the form of bootstrap procedures for assessing reconstruction uncertainty in the real data case. We evaluate the sharpness of this approach and argue that this type of procedure will be critical in the near future when handling the increasing amount of data.