 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQMe14S, A Comprehensive and Efficient Spectral Dataset for Small Organic Molecules
Jan 31, 2025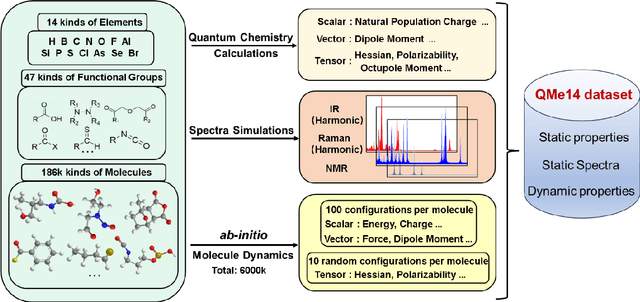
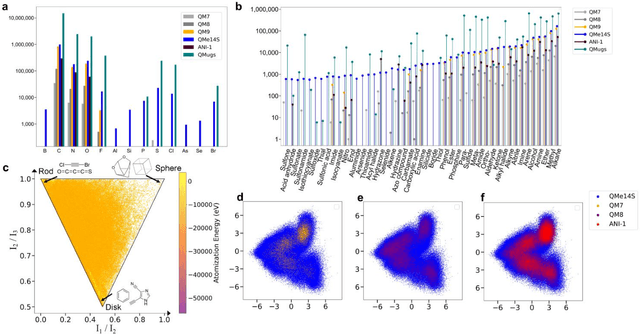
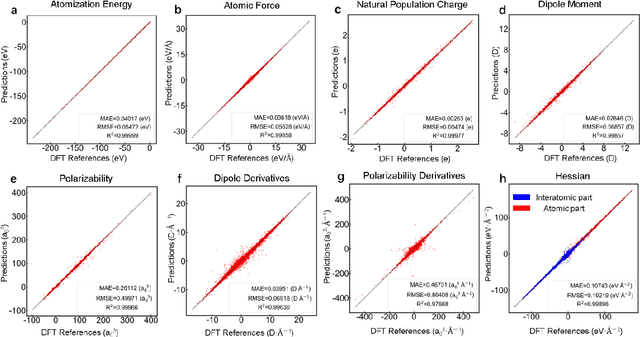

Developing machine learning protocols for molecular simulations requires comprehensive and efficient datasets. Here we introduce the QMe14S dataset, comprising 186,102 small organic molecules featuring 14 elements (H, B, C, N, O, F, Al, Si, P, S, Cl, As, Se, Br) and 47 functional groups. Using density functional theory at the B3LYP/TZVP level, we optimized the geometries and calculated properties including energy, atomic charge, atomic force, dipole moment, quadrupole moment, polarizability, octupole moment, first hyperpolarizability, and Hessian. At the same level, we obtained the harmonic IR, Raman and NMR spectra. Furthermore, we conducted ab initio molecular dynamics simulations to generate dynamic configurations and extract nonequilibrium properties, including energy, forces, and Hessians. By leveraging our E(3)-equivariant message-passing neural network (DetaNet), we demonstrated that models trained on QMe14S outperform those trained on the previously developed QM9S dataset in simulating molecular spectra. The QMe14S dataset thus serves as a comprehensive benchmark for molecular simulations, offering valuable insights into structure-property relationships.