 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeActive learning for affinity prediction of antibodies
Jun 11, 2024

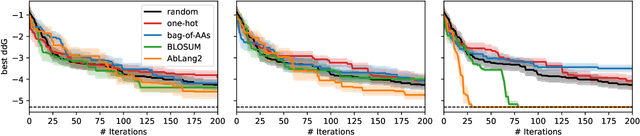

The primary objective of most lead optimization campaigns is to enhance the binding affinity of ligands. For large molecules such as antibodies, identifying mutations that enhance antibody affinity is particularly challenging due to the combinatorial explosion of potential mutations. When the structure of the antibody-antigen complex is available, relative binding free energy (RBFE) methods can offer valuable insights into how different mutations will impact the potency and selectivity of a drug candidate, thereby reducing the reliance on costly and time-consuming wet-lab experiments. However, accurately simulating the physics of large molecules is computationally intensive. We present an active learning framework that iteratively proposes promising sequences for simulators to evaluate, thereby accelerating the search for improved binders. We explore different modeling approaches to identify the most effective surrogate model for this task, and evaluate our framework both using pre-computed pools of data and in a realistic full-loop setting.