 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeep Learning-Based Discrete Calibrated Survival Prediction
Aug 17, 2022
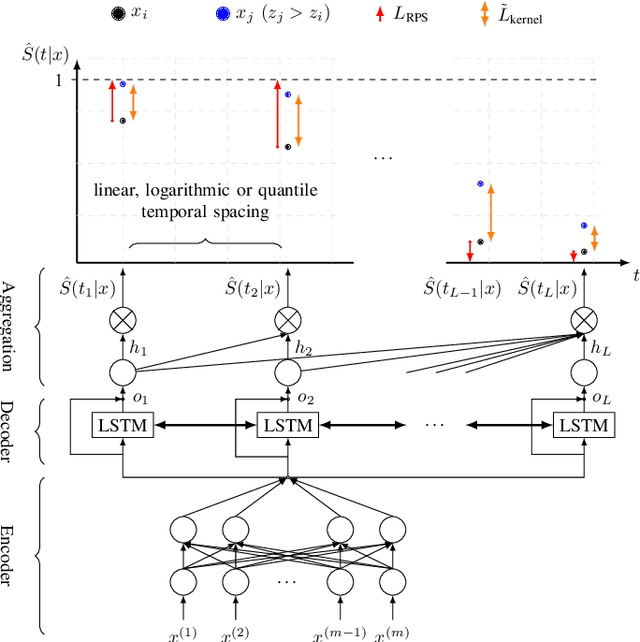

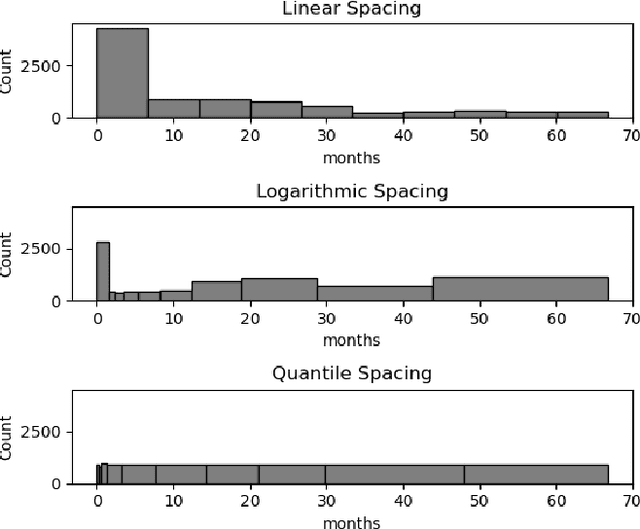
Deep neural networks for survival prediction outper-form classical approaches in discrimination, which is the ordering of patients according to their time-of-event. Conversely, classical approaches like the Cox Proportional Hazards model display much better calibration, the correct temporal prediction of events of the underlying distribution. Especially in the medical domain, where it is critical to predict the survival of a single patient, both discrimination and calibration are important performance metrics. Here we present Discrete Calibrated Survival (DCS), a novel deep neural network for discriminated and calibrated survival prediction that outperforms competing survival models in discrimination on three medical datasets, while achieving best calibration among all discrete time models. The enhanced performance of DCS can be attributed to two novel features, the variable temporal output node spacing and the novel loss term that optimizes the use of uncensored and censored patient data. We believe that DCS is an important step towards clinical application of deep-learning-based survival prediction with state-of-the-art discrimination and good calibration.
Towards Explainable End-to-End Prostate Cancer Relapse Prediction from H&E Images Combining Self-Attention Multiple Instance Learning with a Recurrent Neural Network
Nov 26, 2021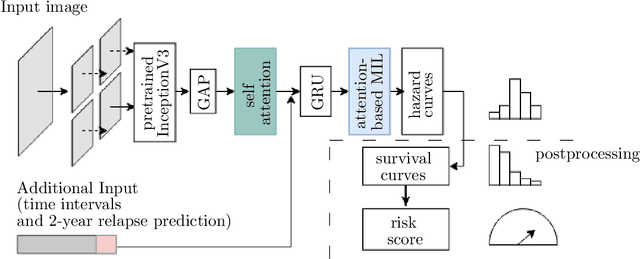
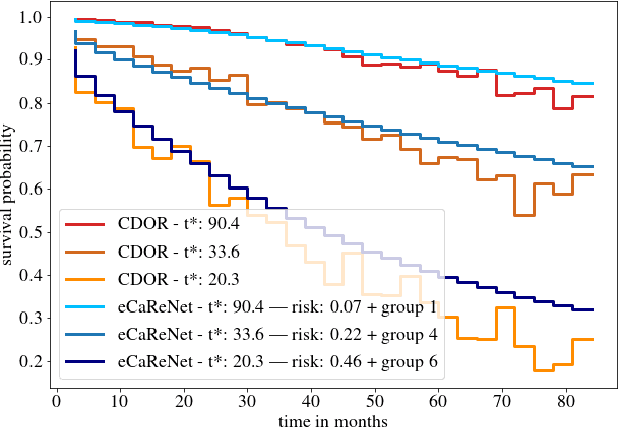
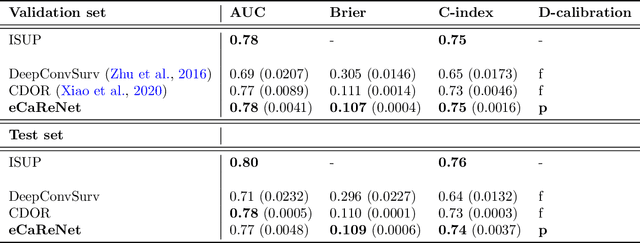
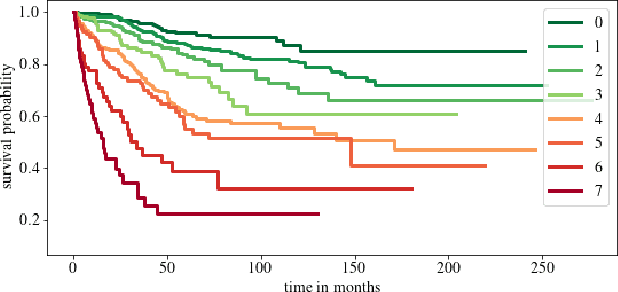
Clinical decision support for histopathology image data mainly focuses on strongly supervised annotations, which offers intuitive interpretability, but is bound by expert performance. Here, we propose an explainable cancer relapse prediction network (eCaReNet) and show that end-to-end learning without strong annotations offers state-of-the-art performance while interpretability can be included through an attention mechanism. On the use case of prostate cancer survival prediction, using 14,479 images and only relapse times as annotations, we reach a cumulative dynamic AUC of 0.78 on a validation set, being on par with an expert pathologist (and an AUC of 0.77 on a separate test set). Our model is well-calibrated and outputs survival curves as well as a risk score and group per patient. Making use of the attention weights of a multiple instance learning layer, we show that malignant patches have a higher influence on the prediction than benign patches, thus offering an intuitive interpretation of the prediction. Our code is available at www.github.com/imsb-uke/ecarenet.
Deep learning-based bias transfer for overcoming laboratory differences of microscopic images
May 25, 2021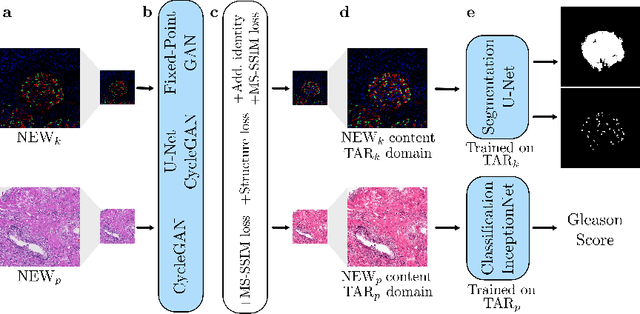
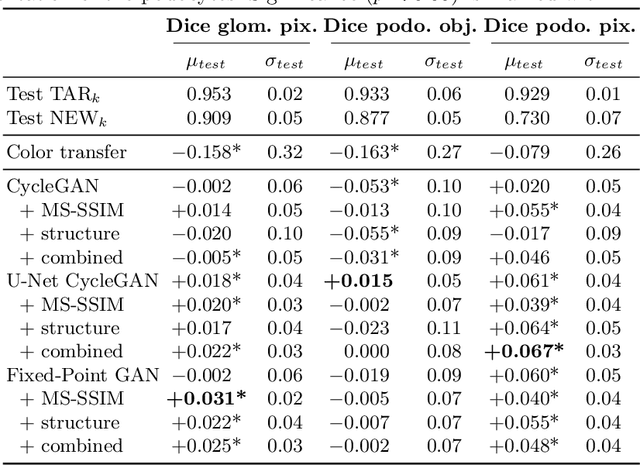
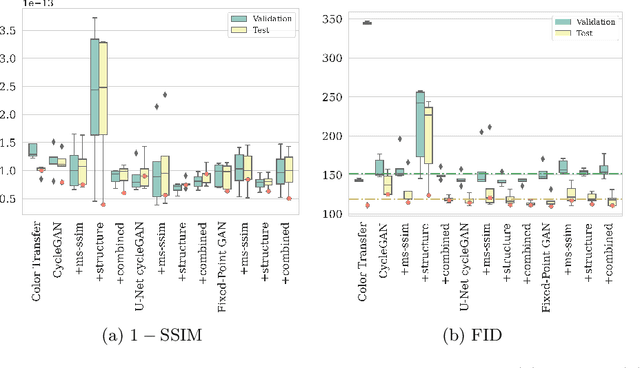
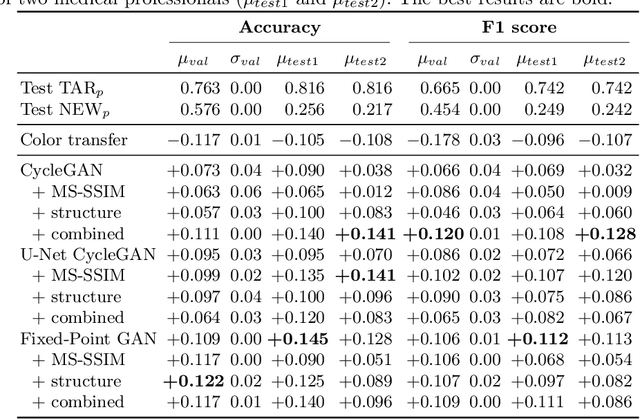
The automated analysis of medical images is currently limited by technical and biological noise and bias. The same source tissue can be represented by vastly different images if the image acquisition or processing protocols vary. For an image analysis pipeline, it is crucial to compensate such biases to avoid misinterpretations. Here, we evaluate, compare, and improve existing generative model architectures to overcome domain shifts for immunofluorescence (IF) and Hematoxylin and Eosin (H&E) stained microscopy images. To determine the performance of the generative models, the original and transformed images were segmented or classified by deep neural networks that were trained only on images of the target bias. In the scope of our analysis, U-Net cycleGANs trained with an additional identity and an MS-SSIM-based loss and Fixed-Point GANs trained with an additional structure loss led to the best results for the IF and H&E stained samples, respectively. Adapting the bias of the samples significantly improved the pixel-level segmentation for human kidney glomeruli and podocytes and improved the classification accuracy for human prostate biopsies by up to 14%.
Explainable Deep Learning for Augmentation of sRNA Expression Profiles
Sep 26, 2019



The lack of well-structured metadata annotations complicates there-usability and interpretation of the growing amount of publicly available RNA expression data. The machine learning-based prediction of metadata(data augmentation) can considerably improve the quality of expression data annotation. In this study,we systematically benchmark deep learning (DL) and random forest (RF)-based metadata augmentation of tissue, age, and sex using small RNA (sRNA) expression profiles. We use 4243 annotated sRNA-Seq samples from the small RNA expression atlas (SEA) database to train and test the augmentation performance. In general, the DL machine learner outperforms the RF method in almost all tested cases. The average cross-validated prediction accuracy of the DL algorithm for tissues is 96.5%, for sex is 77%, and for age is 77.2%. The average tissue prediction accuracy for a completely new dataset is 83.1% (DL) and 80.8% (RF). To understand which sRNAs influence DL predictions, we employ backpropagation-based feature importance scores using the DeepLIFT method, which enable us to obtain information on biological relevance of sRNAs.
Deep Learning and Random Forest-Based Augmentation of sRNA Expression Profiles
Sep 26, 2019
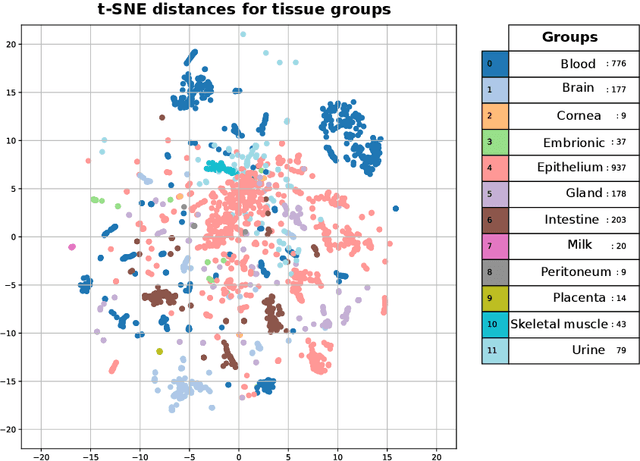

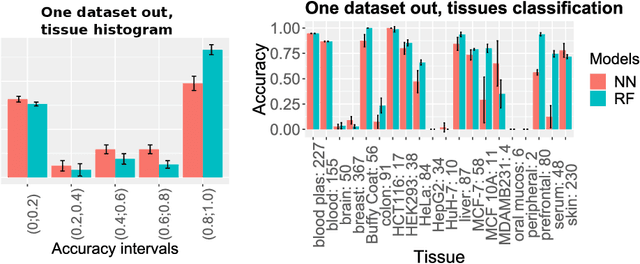
The lack of well-structured annotations in a growing amount of RNA expression data complicates data interoperability and reusability. Commonly - used text mining methods extract annotations from existing unstructured data descriptions and often provide inaccurate output that requires manual curation. Automatic data-based augmentation (generation of annotations on the base of expression data) can considerably improve the annotation quality and has not been well-studied. We formulate an automatic augmentation of small RNA-seq expression data as a classification problem and investigate deep learning (DL) and random forest (RF) approaches to solve it. We generate tissue and sex annotations from small RNA-seq expression data for tissues and cell lines of homo sapiens. We validate our approach on 4243 annotated small RNA-seq samples from the Small RNA Expression Atlas (SEA) database. The average prediction accuracy for tissue groups is 98% (DL), for tissues - 96.5% (DL), and for sex - 77% (DL). The "one dataset out" average accuracy for tissue group prediction is 83% (DL) and 59% (RF). On average, DL provides better results as compared to RF, and considerably improves classification performance for 'unseen' datasets.