 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInvestigating Knowledge Distillation Through Neural Networks for Protein Binding Affinity Prediction
Jan 07, 2026The trade-off between predictive accuracy and data availability makes it difficult to predict protein--protein binding affinity accurately. The lack of experimentally resolved protein structures limits the performance of structure-based machine learning models, which generally outperform sequence-based methods. In order to overcome this constraint, we suggest a regression framework based on knowledge distillation that uses protein structural data during training and only needs sequence data during inference. The suggested method uses binding affinity labels and intermediate feature representations to jointly supervise the training of a sequence-based student network under the guidance of a structure-informed teacher network. Leave-One-Complex-Out (LOCO) cross-validation was used to assess the framework on a non-redundant protein--protein binding affinity benchmark dataset. A maximum Pearson correlation coefficient (P_r) of 0.375 and an RMSE of 2.712 kcal/mol were obtained by sequence-only baseline models, whereas a P_r of 0.512 and an RMSE of 2.445 kcal/mol were obtained by structure-based models. With a P_r of 0.481 and an RMSE of 2.488 kcal/mol, the distillation-based student model greatly enhanced sequence-only performance. Improved agreement and decreased bias were further confirmed by thorough error analyses. With the potential to close the performance gap between sequence-based and structure-based models as larger datasets become available, these findings show that knowledge distillation is an efficient method for transferring structural knowledge to sequence-based predictors. The source code for running inference with the proposed distillation-based binding affinity predictor can be accessed at https://github.com/wajidarshad/ProteinAffinityKD.
COVIDX: Computer-aided diagnosis of Covid-19 and its severity prediction with raw digital chest X-ray images
Dec 25, 2020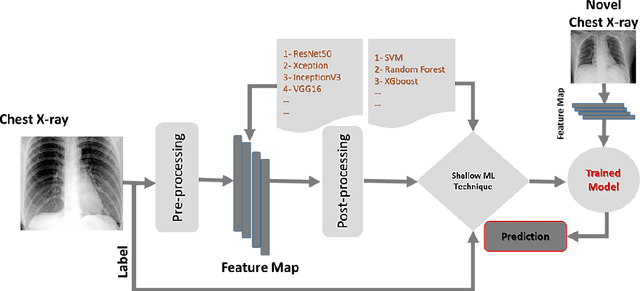
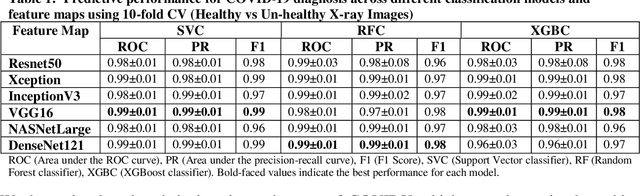

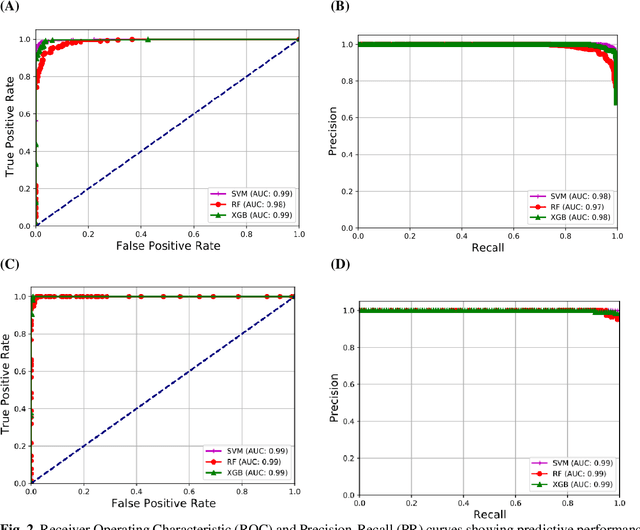
Coronavirus disease (COVID-19) is a contagious infection caused by severe acute respiratory syndrome coronavirus-2 (SARS-COV-2) and it has infected and killed millions of people across the globe. In the absence of specific drugs or vaccines for the treatment of COVID-19 and the limitation of prevailing diagnostic techniques, there is a requirement for some alternate automatic screening systems that can be used by the physicians to quickly identify and isolate the infected patients. A chest X-ray (CXR) image can be used as an alternative modality to detect and diagnose the COVID-19. In this study, we present an automatic COVID-19 diagnostic and severity prediction (COVIDX) system that uses deep feature maps from CXR images to diagnose COVID-19 and its severity prediction. The proposed system uses a three-phase classification approach (healthy vs unhealthy, COVID-19 vs Pneumonia, and COVID-19 severity) using different shallow supervised classification algorithms. We evaluated COVIDX not only through 10-fold cross2 validation and by using an external validation dataset but also in real settings by involving an experienced radiologist. In all the evaluation settings, COVIDX outperforms all the existing stateof-the-art methods designed for this purpose. We made COVIDX easily accessible through a cloud-based webserver and python code available at https://sites.google.com/view/wajidarshad/software and https://github.com/wajidarshad/covidx, respectively.
PANDA: Predicting the change in proteins binding affinity upon mutations using sequence information
Sep 16, 2020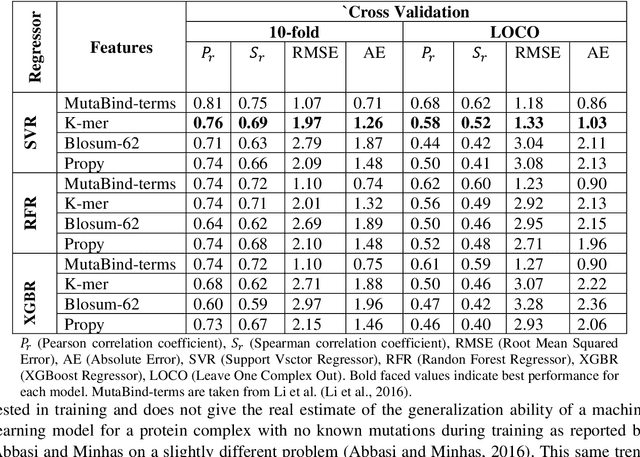

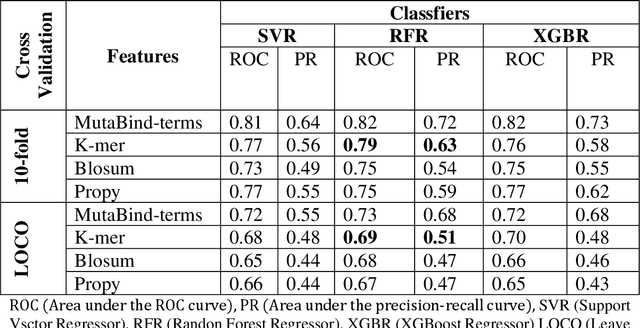
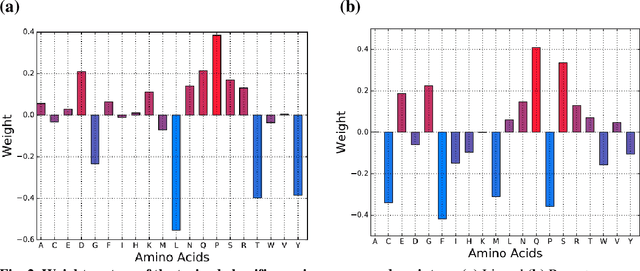
Accurately determining a change in protein binding affinity upon mutations is important for the discovery and design of novel therapeutics and to assist mutagenesis studies. Determination of change in binding affinity upon mutations requires sophisticated, expensive, and time-consuming wet-lab experiments that can be aided with computational methods. Most of the computational prediction techniques require protein structures that limit their applicability to protein complexes with known structures. In this work, we explore the sequence-based prediction of change in protein binding affinity upon mutation. We have used protein sequence information instead of protein structures along with machine learning techniques to accurately predict the change in protein binding affinity upon mutation. Our proposed sequence-based novel change in protein binding affinity predictor called PANDA gives better accuracy than existing methods over the same validation set as well as on an external independent test dataset. On an external test dataset, our proposed method gives a maximum Pearson correlation coefficient of 0.52 in comparison to the state-of-the-art existing protein structure-based method called MutaBind which gives a maximum Pearson correlation coefficient of 0.59. Our proposed protein sequence-based method, to predict a change in binding affinity upon mutations, has wide applicability and comparable performance in comparison to existing protein structure-based methods. A cloud-based webserver implementation of PANDA and its python code is available at https://sites.google.com/view/wajidarshad/software and https://github.com/wajidarshad/panda.