 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSemi-Supervised Quantile Estimation: Robust and Efficient Inference in High Dimensional Settings
Jan 25, 2022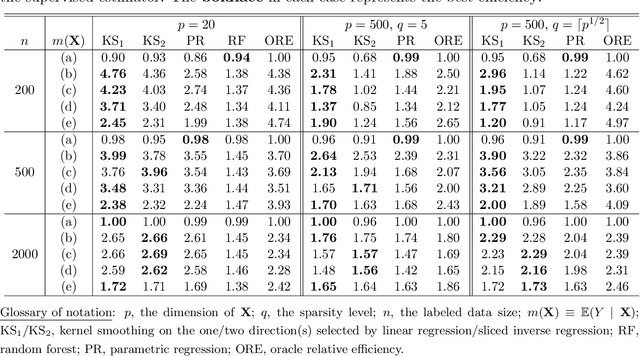
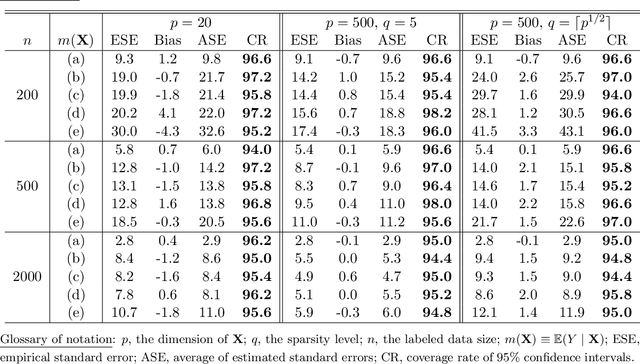
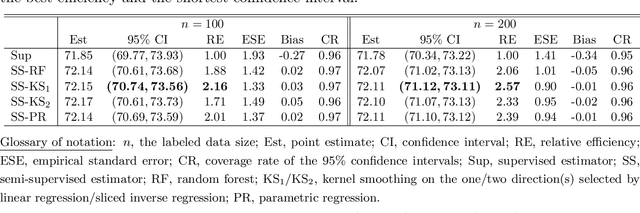
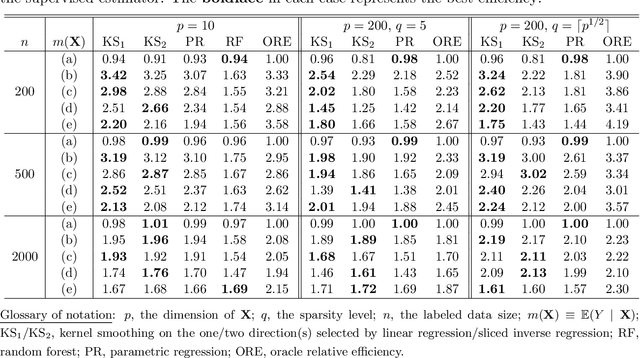
We consider quantile estimation in a semi-supervised setting, characterized by two available data sets: (i) a small or moderate sized labeled data set containing observations for a response and a set of possibly high dimensional covariates, and (ii) a much larger unlabeled data set where only the covariates are observed. We propose a family of semi-supervised estimators for the response quantile(s) based on the two data sets, to improve the estimation accuracy compared to the supervised estimator, i.e., the sample quantile from the labeled data. These estimators use a flexible imputation strategy applied to the estimating equation along with a debiasing step that allows for full robustness against misspecification of the imputation model. Further, a one-step update strategy is adopted to enable easy implementation of our method and handle the complexity from the non-linear nature of the quantile estimating equation. Under mild assumptions, our estimators are fully robust to the choice of the nuisance imputation model, in the sense of always maintaining root-n consistency and asymptotic normality, while having improved efficiency relative to the supervised estimator. They also attain semi-parametric optimality if the relation between the response and the covariates is correctly specified via the imputation model. As an illustration of estimating the nuisance imputation function, we consider kernel smoothing type estimators on lower dimensional and possibly estimated transformations of the high dimensional covariates, and we establish novel results on their uniform convergence rates in high dimensions, involving responses indexed by a function class and usage of dimension reduction techniques. These results may be of independent interest. Numerical results on both simulated and real data confirm our semi-supervised approach's improved performance, in terms of both estimation and inference.
Data Integration with High Dimensionality
Oct 03, 2016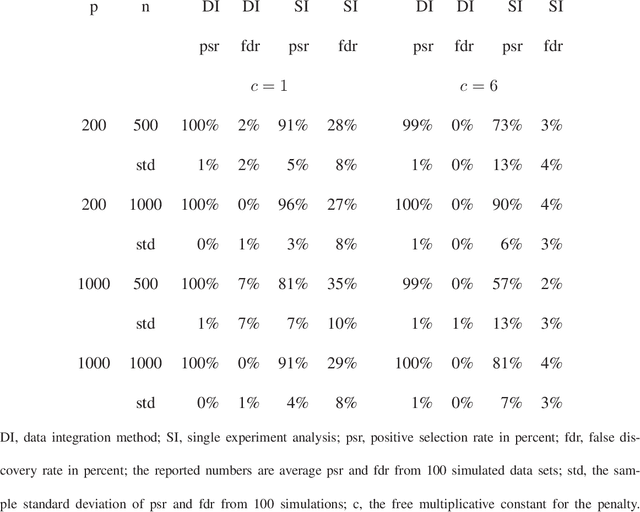
We consider a problem of data integration. Consider determining which genes affect a disease. The genes, which we call predictor objects, can be measured in different experiments on the same individual. We address the question of finding which genes are predictors of disease by any of the experiments. Our formulation is more general. In a given data set, there are a fixed number of responses for each individual, which may include a mix of discrete, binary and continuous variables. There is also a class of predictor objects, which may differ within a subject depending on how the predictor object is measured, i.e., depend on the experiment. The goal is to select which predictor objects affect any of the responses, where the number of such informative predictor objects or features tends to infinity as sample size increases. There are marginal likelihoods for each way the predictor object is measured, i.e., for each experiment. We specify a pseudolikelihood combining the marginal likelihoods, and propose a pseudolikelihood information criterion. Under regularity conditions, we establish selection consistency for the pseudolikelihood information criterion with unbounded true model size, which includes a Bayesian information criterion with appropriate penalty term as a special case. Simulations indicate that data integration improves upon, sometimes dramatically, using only one of the data sources.
Use of multiple singular value decompositions to analyze complex intracellular calcium ion signals
Sep 28, 2010

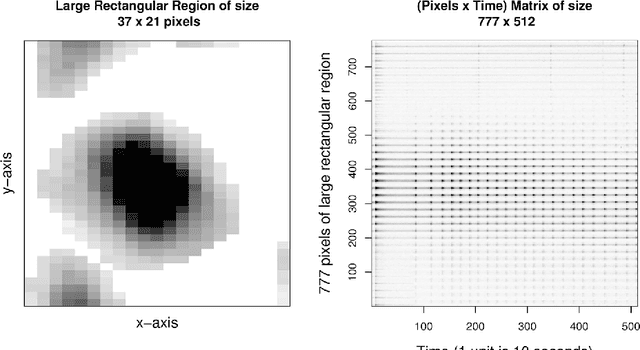

We compare calcium ion signaling ($\mathrm {Ca}^{2+}$) between two exposures; the data are present as movies, or, more prosaically, time series of images. This paper describes novel uses of singular value decompositions (SVD) and weighted versions of them (WSVD) to extract the signals from such movies, in a way that is semi-automatic and tuned closely to the actual data and their many complexities. These complexities include the following. First, the images themselves are of no interest: all interest focuses on the behavior of individual cells across time, and thus, the cells need to be segmented in an automated manner. Second, the cells themselves have 100$+$ pixels, so that they form 100$+$ curves measured over time, so that data compression is required to extract the features of these curves. Third, some of the pixels in some of the cells are subject to image saturation due to bit depth limits, and this saturation needs to be accounted for if one is to normalize the images in a reasonably unbiased manner. Finally, the $\mathrm {Ca}^{2+}$ signals have oscillations or waves that vary with time and these signals need to be extracted. Thus, our aim is to show how to use multiple weighted and standard singular value decompositions to detect, extract and clarify the $\mathrm {Ca}^{2+}$ signals. Our signal extraction methods then lead to simple although finely focused statistical methods to compare $\mathrm {Ca}^{2+}$ signals across experimental conditions.
* Published in at http://dx.doi.org/10.1214/09-AOAS253 the Annals of Applied Statistics (http://www.imstat.org/aoas/) by the Institute of Mathematical Statistics (http://www.imstat.org)