 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeValue-Added Chemical Discovery Using Reinforcement Learning
Nov 10, 2019
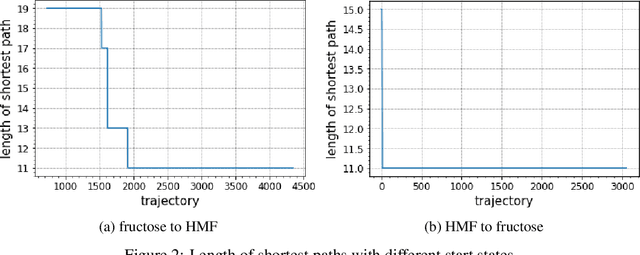

Computer-assisted synthesis planning aims to help chemists find better reaction pathways faster. Finding viable and short pathways from sugar molecules to value-added chemicals can be modeled as a retrosynthesis planning problem with a catalyst allowed. This is a crucial step in efficient biomass conversion. The traditional computational chemistry approach to identifying possible reaction pathways involves computing the reaction energies of hundreds of intermediates, which is a critical bottleneck in silico reaction discovery. Deep reinforcement learning has shown in other domains that a well-trained agent with little or no prior human knowledge can surpass human performance. While some effort has been made to adapt machine learning techniques to the retrosynthesis planning problem, value-added chemical discovery presents unique challenges. Specifically, the reaction can occur in several different sites in a molecule, a subtle case that has never been treated in previous works. With a more versatile formulation of the problem as a Markov decision process, we address the problem using deep reinforcement learning techniques and present promising preliminary results.