 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIterative Peptide Modeling With Active Learning And Meta-Learning
Nov 20, 2019



Often the development of novel materials is not amenable to high-throughput or purely computational screening methods. Instead, materials must be synthesized one at a time in a process that does not generate significant amounts of data. One way this method can be improved is by ensuring that each experiment provides the best improvement in both material properties and predictive modeling accuracy. In this work, we study the effectiveness of active learning, which optimizes the order of experiments, and meta learning, which transfers knowledge from one context to another, to reduce the number of experiments necessary to build a predictive model. We present a novel multi-task benchmark database of peptides designed to advance active, few-shot, and meta-learning methods for experimental design. Each task is binary classification of peptides represented as a sequence string. We show results of standard active learning and meta-learning methods across these datasets to assess their ability to improve predictive models with the fewest number of experiments. We find the ensemble query by committee active learning method to be effective. The meta-learning method Reptile was found to improve accuracy. The robustness of these conclusions were tested across multiple model choices.
Classifying Antimicrobial and Multifunctional Peptides with Bayesian Network Models
Apr 17, 2018
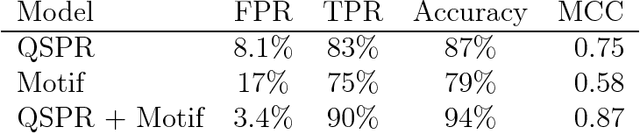


Bayesian network models are finding success in characterizing enzyme-catalyzed reactions, slow conformational changes, predicting enzyme inhibition, and genomics. In this work, we apply them to statistical modeling of peptides by simultaneously identifying amino acid sequence motifs and using a motif-based model to clarify the role motifs may play in antimicrobial activity. We construct models of increasing sophistication, demonstrating how chemical knowledge of a peptide system may be embedded without requiring new derivation of model fitting equations after changing model structure. These models are used to construct classifiers with good performance (94% accuracy, Matthews correlation coefficient of 0.87) at predicting antimicrobial activity in peptides, while at the same time being built of interpretable parameters. We demonstrate use of these models to identify peptides that are potentially both antimicrobial and antifouling, and show that the background distribution of amino acids could play a greater role in activity than sequence motifs do. This provides an advancement in the type of peptide activity modeling that can be done and the ease in which models can be constructed.
* 19 pages, 7 figures, 1 table, supporting information included