 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRe-parameterizing VAEs for stability
Jun 25, 2021
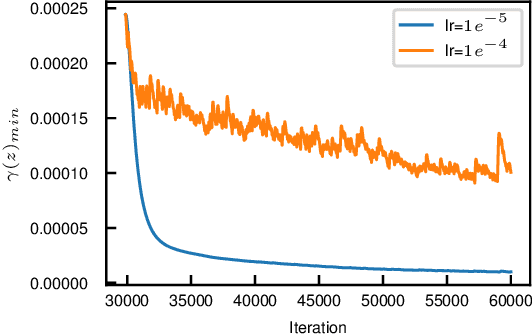
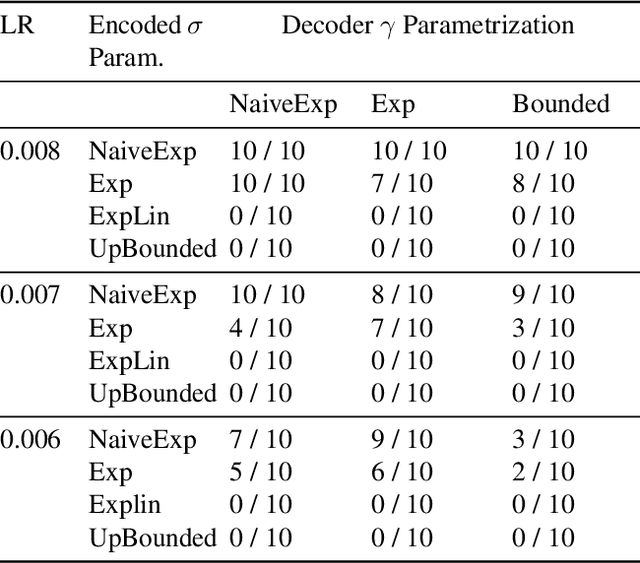
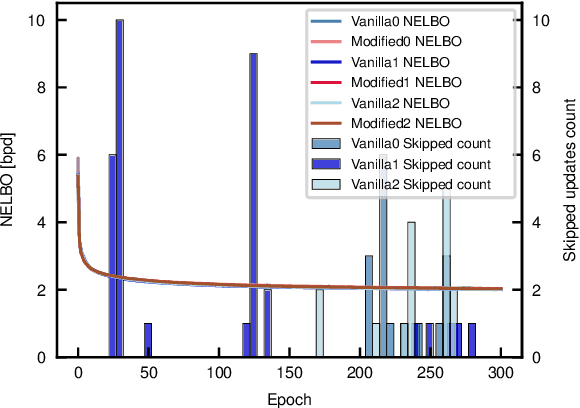
We propose a theoretical approach towards the training numerical stability of Variational AutoEncoders (VAE). Our work is motivated by recent studies empowering VAEs to reach state of the art generative results on complex image datasets. These very deep VAE architectures, as well as VAEs using more complex output distributions, highlight a tendency to haphazardly produce high training gradients as well as NaN losses. The empirical fixes proposed to train them despite their limitations are neither fully theoretically grounded nor generally sufficient in practice. Building on this, we localize the source of the problem at the interface between the model's neural networks and their output probabilistic distributions. We explain a common source of instability stemming from an incautious formulation of the encoded Normal distribution's variance, and apply the same approach on other, less obvious sources. We show that by implementing small changes to the way we parameterize the Normal distributions on which they rely, VAEs can securely be trained.
Graph Context Encoder: Graph Feature Inpainting for Graph Generation and Self-supervised Pretraining
Jun 18, 2021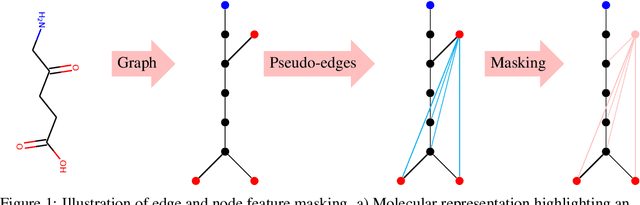
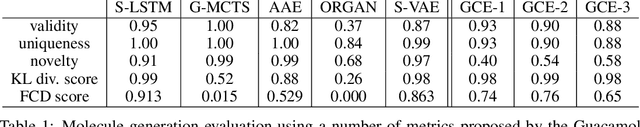
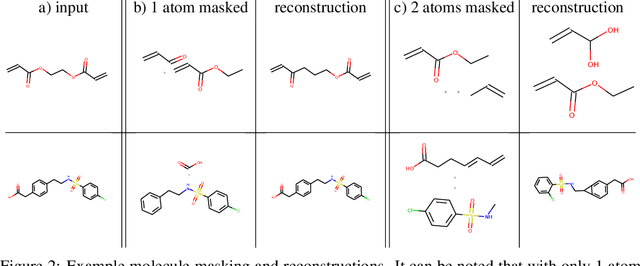

We propose the Graph Context Encoder (GCE), a simple but efficient approach for graph representation learning based on graph feature masking and reconstruction. GCE models are trained to efficiently reconstruct input graphs similarly to a graph autoencoder where node and edge labels are masked. In particular, our model is also allowed to change graph structures by masking and reconstructing graphs augmented by random pseudo-edges. We show that GCE can be used for novel graph generation, with applications for molecule generation. Used as a pretraining method, we also show that GCE improves baseline performances in supervised classification tasks tested on multiple standard benchmark graph datasets.
Realistic molecule optimization on a learned graph manifold
Jun 03, 2021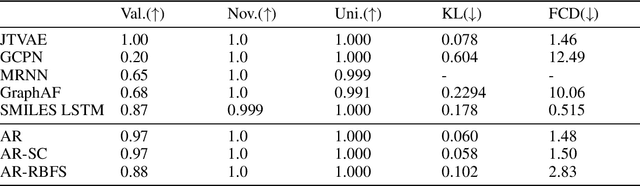
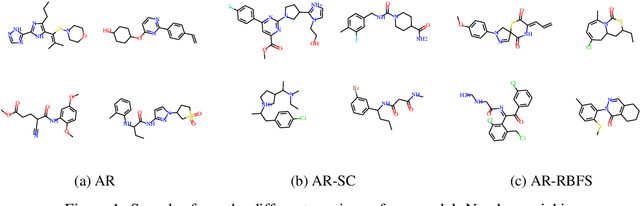
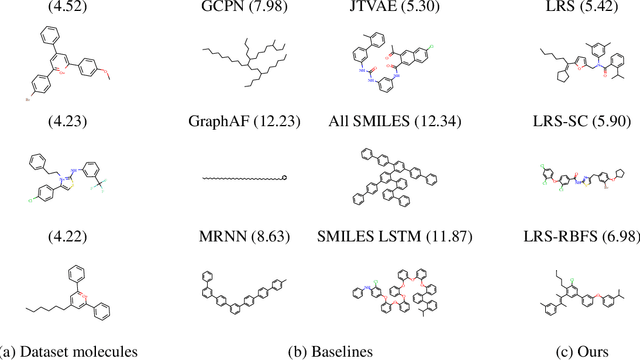
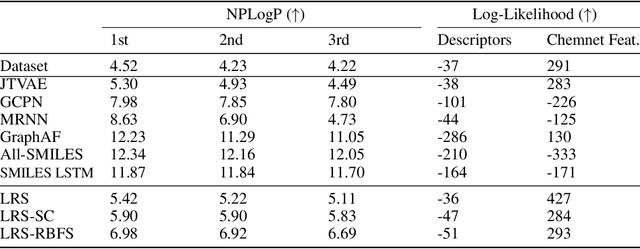
Deep learning based molecular graph generation and optimization has recently been attracting attention due to its great potential for de novo drug design. On the one hand, recent models are able to efficiently learn a given graph distribution, and many approaches have proven very effective to produce a molecule that maximizes a given score. On the other hand, it was shown by previous studies that generated optimized molecules are often unrealistic, even with the inclusion of mechanics to enforce similarity to a dataset of real drug molecules. In this work we use a hybrid approach, where the dataset distribution is learned using an autoregressive model while the score optimization is done using the Metropolis algorithm, biased toward the learned distribution. We show that the resulting method, that we call learned realism sampling (LRS), produces empirically more realistic molecules and outperforms all recent baselines in the task of molecule optimization with similarity constraints.
Graph convolutions that can finally model local structure
Nov 30, 2020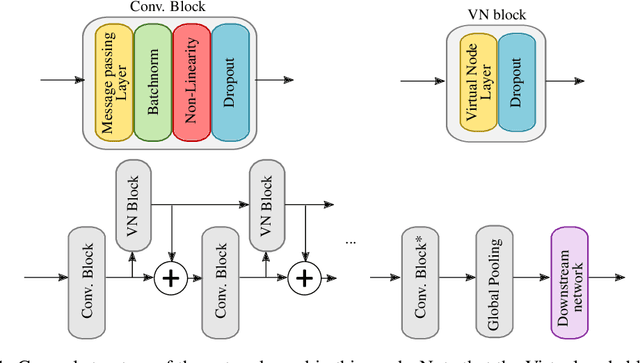
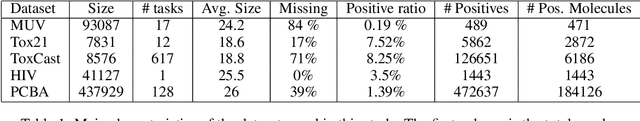
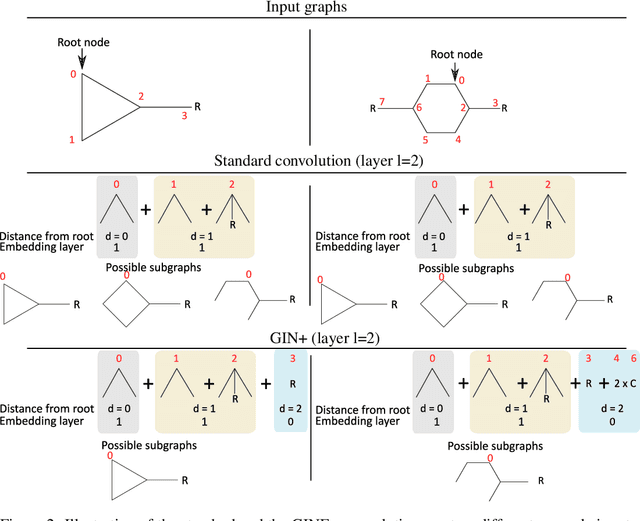
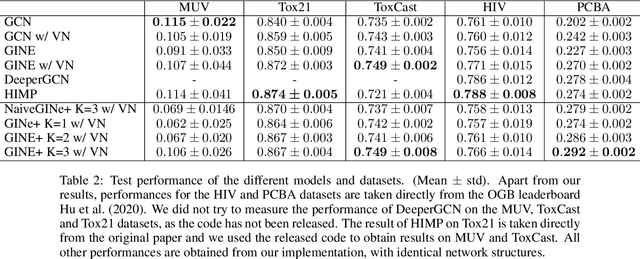
Despite quick progress in the last few years, recent studies have shown that modern graph neural networks can still fail at very simple tasks, like detecting small cycles. This hints at the fact that current networks fail to catch information about the local structure, which is problematic if the downstream task heavily relies on graph substructure analysis, as in the context of chemistry. We propose a very simple correction to the now standard GIN convolution that enables the network to detect small cycles with nearly no cost in terms of computation time and number of parameters. Tested on real life molecule property datasets, our model consistently improves performance on large multi-tasked datasets over all baselines, both globally and on a per-task setting.