 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRealistic molecule optimization on a learned graph manifold
Paper and Code
Jun 03, 2021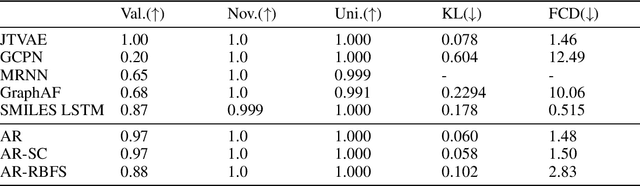
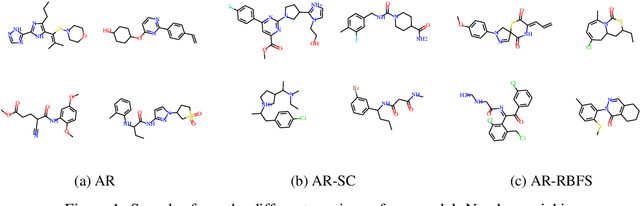
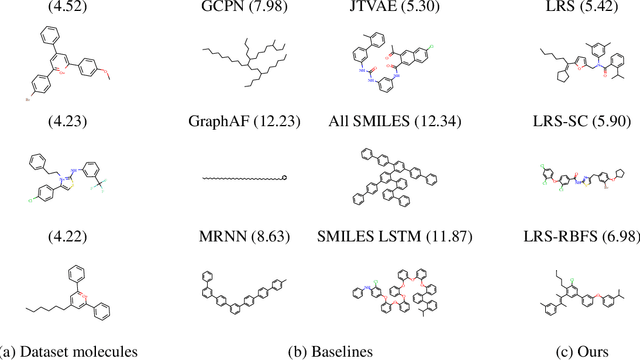
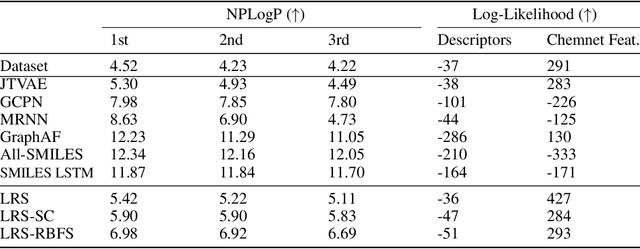
Deep learning based molecular graph generation and optimization has recently been attracting attention due to its great potential for de novo drug design. On the one hand, recent models are able to efficiently learn a given graph distribution, and many approaches have proven very effective to produce a molecule that maximizes a given score. On the other hand, it was shown by previous studies that generated optimized molecules are often unrealistic, even with the inclusion of mechanics to enforce similarity to a dataset of real drug molecules. In this work we use a hybrid approach, where the dataset distribution is learned using an autoregressive model while the score optimization is done using the Metropolis algorithm, biased toward the learned distribution. We show that the resulting method, that we call learned realism sampling (LRS), produces empirically more realistic molecules and outperforms all recent baselines in the task of molecule optimization with similarity constraints.