 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCryoLVM: Self-supervised Learning from Cryo-EM Density Maps with Large Vision Models
Feb 02, 2026Cryo-electron microscopy (cryo-EM) has revolutionized structural biology by enabling near-atomic-level visualization of biomolecular assemblies. However, the exponential growth in cryo-EM data throughput and complexity, coupled with diverse downstream analytical tasks, necessitates unified computational frameworks that transcend current task-specific deep learning approaches with limited scalability and generalizability. We present CryoLVM, a foundation model that learns rich structural representations from experimental density maps with resolved structures by leveraging the Joint-Embedding Predictive Architecture (JEPA) integrated with SCUNet-based backbone, which can be rapidly adapted to various downstream tasks. We further introduce a novel histogram-based distribution alignment loss that accelerates convergence and enhances fine-tuning performance. We demonstrate CryoLVM's effectiveness across three critical cryo-EM tasks: density map sharpening, density map super-resolution, and missing wedge restoration. Our method consistently outperforms state-of-the-art baselines across multiple density map quality metrics, confirming its potential as a versatile model for a wide spectrum of cryo-EM applications.
A^2-Net: Molecular Structure Estimation from Cryo-EM Density Volumes
Jan 06, 2019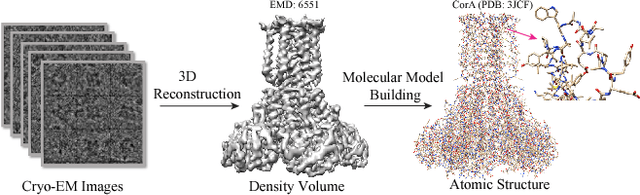



Constructing of molecular structural models from Cryo-Electron Microscopy (Cryo-EM) density volumes is the critical last step of structure determination by Cryo-EM technologies. Methods have evolved from manual construction by structural biologists to perform 6D translation-rotation searching, which is extremely compute-intensive. In this paper, we propose a learning-based method and formulate this problem as a vision-inspired 3D detection and pose estimation task. We develop a deep learning framework for amino acid determination in a 3D Cryo-EM density volume. We also design a sequence-guided Monte Carlo Tree Search (MCTS) to thread over the candidate amino acids to form the molecular structure. This framework achieves 91% coverage on our newly proposed dataset and takes only a few minutes for a typical structure with a thousand amino acids. Our method is hundreds of times faster and several times more accurate than existing automated solutions without any human intervention.
* 8 pages, 5 figures, 4 tables