 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePlug & Play Directed Evolution of Proteins with Gradient-based Discrete MCMC
Dec 20, 2022A long-standing goal of machine-learning-based protein engineering is to accelerate the discovery of novel mutations that improve the function of a known protein. We introduce a sampling framework for evolving proteins in silico that supports mixing and matching a variety of unsupervised models, such as protein language models, and supervised models that predict protein function from sequence. By composing these models, we aim to improve our ability to evaluate unseen mutations and constrain search to regions of sequence space likely to contain functional proteins. Our framework achieves this without any model fine-tuning or re-training by constructing a product of experts distribution directly in discrete protein space. Instead of resorting to brute force search or random sampling, which is typical of classic directed evolution, we introduce a fast MCMC sampler that uses gradients to propose promising mutations. We conduct in silico directed evolution experiments on wide fitness landscapes and across a range of different pre-trained unsupervised models, including a 650M parameter protein language model. Our results demonstrate an ability to efficiently discover variants with high evolutionary likelihood as well as estimated activity multiple mutations away from a wild type protein, suggesting our sampler provides a practical and effective new paradigm for machine-learning-based protein engineering.
Message-passing neural networks for high-throughput polymer screening
Jul 26, 2018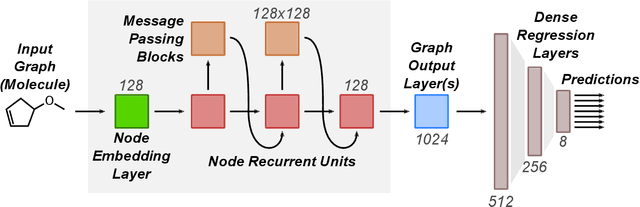
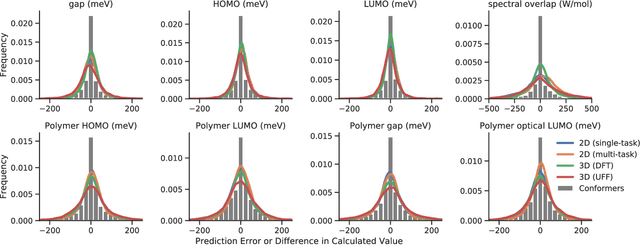
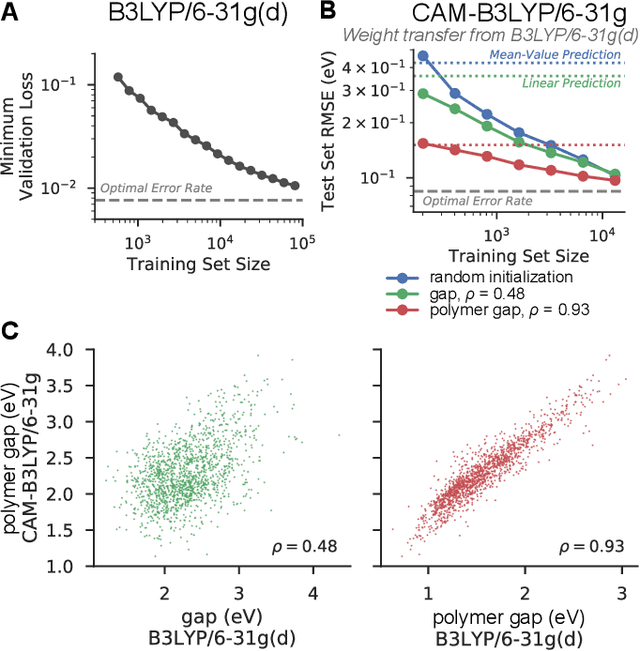
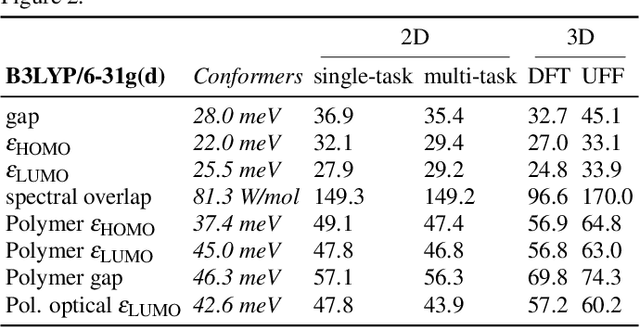
Machine learning methods have shown promise in predicting molecular properties, and given sufficient training data ML may surpass density functional theory in computational speed and chemical accuracy. However, the most accurate machine learning methods require optimized 3D molecular geometries, limiting their applicability for high-throughput screening. We show that near-optimal results for large polymeric molecules can be obtained without optimized 3D geometry, and that trained model weights can be used to improve performance on related tasks.